- Retinitis pigmentosa

Содержание
- 2. En gruppe av arvebetinget sykdommer (retinale PE dystrofier) som er preget med progressiv bilateral perifer visustap
- 3. Genetisk determinasjon fører til økende apoptose i PE celler (der finns det tapper=cone=iodopsiner og staver=rod=rhodopisner som
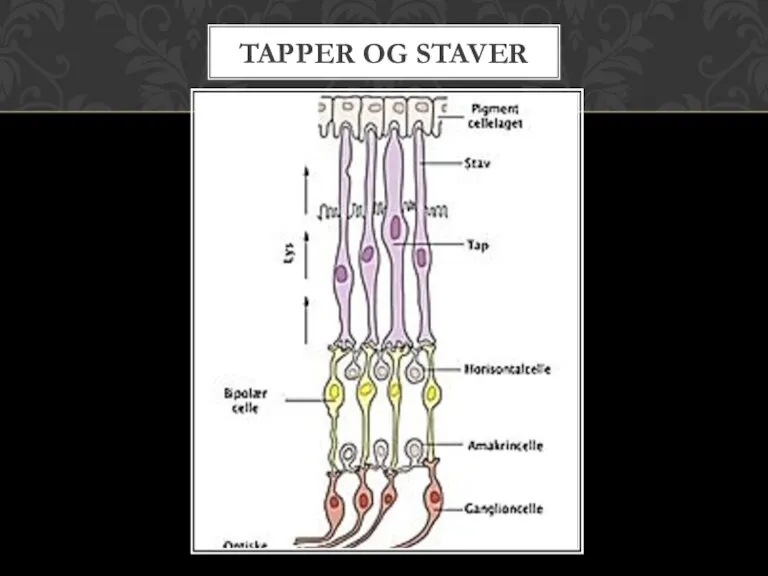
- 4. TAPPER OG STAVER
- 5. I hele verden 1/5000, 1/4000 (USA) Høyest blant indianere i Navajo 1/1878 Lavest 1/7000 i Sweitz
- 6. Nyctalopia Sakte progredierende visus tap først perifert, senere sentralt også, særlig ved tilfeller med ME Fotopsier,
- 7. Dersom RP assosiert med mange esktraokulære tegn og syndromer, det er først og fremst vanlig klinsik
- 8. Øyeundersøkelse: visus, SF, oftalmoskopi, ERG (Elektroretinogramm), fargetest med oppmerksomhet for blå og gule fargeblindhet, mørkeadaptasjon Bildediagnostikk:
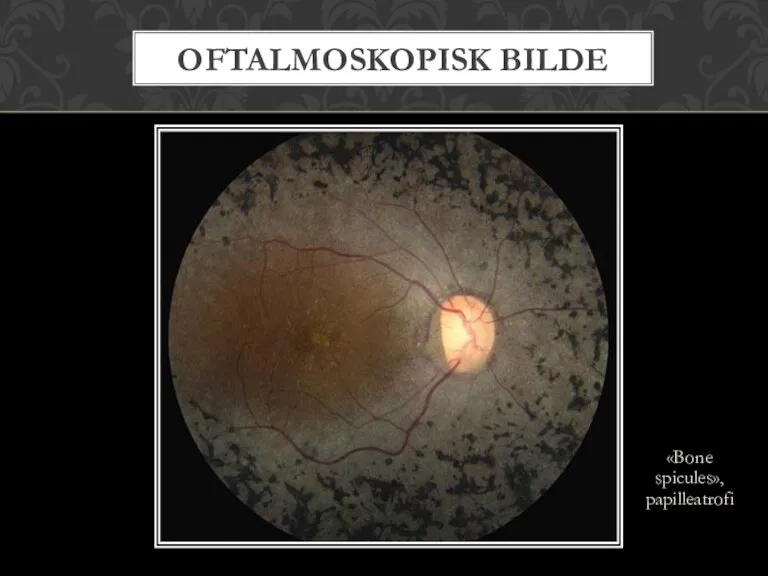
- 9. «Bone spicules», papilleatrofi OFTALMOSKOPISK BILDE
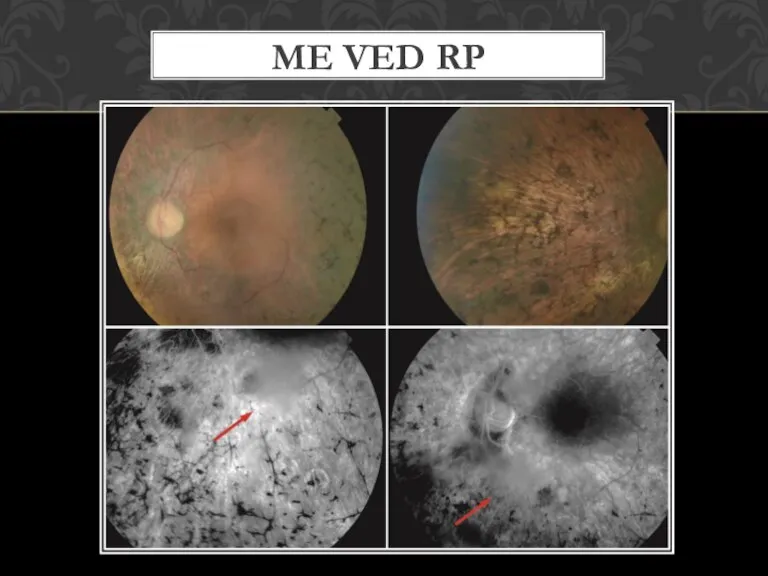
- 10. ME VED RP
- 11. SENTRAL SF VED RP
- 12. Ingen behandling visst seg effektiv Vitamin A og E forsøk i høye doser , ascorbinsyre per
- 13. Prognose er generelt dårlig, sykdom progredierer livet ut Kjønnsbundet former progredierer raskere og gir betydelig visustap
- 15. Скачать презентацию

Слайд 3Genetisk determinasjon fører til økende apoptose i PE celler (der finns det
Genetisk determinasjon fører til økende apoptose i PE celler (der finns det

Слайд 4TAPPER OG STAVER
TAPPER OG STAVER
Слайд 5I hele verden 1/5000, 1/4000 (USA)
Høyest blant indianere i Navajo 1/1878
Lavest
I hele verden 1/5000, 1/4000 (USA)
Høyest blant indianere i Navajo 1/1878
Lavest
Høyest blant indianere i Navajo 1/1878
Lavest

Слайд 6Nyctalopia
Sakte progredierende visus tap først perifert, senere sentralt også, særlig ved tilfeller
Nyctalopia
Sakte progredierende visus tap først perifert, senere sentralt også, særlig ved tilfeller
Sakte progredierende visus tap først perifert, senere sentralt også, særlig ved tilfeller
Слайд 7Dersom RP assosiert med mange esktraokulære tegn og syndromer, det er først
Dersom RP assosiert med mange esktraokulære tegn og syndromer, det er først

Слайд 8Øyeundersøkelse: visus, SF, oftalmoskopi, ERG (Elektroretinogramm), fargetest med oppmerksomhet for blå og
Øyeundersøkelse: visus, SF, oftalmoskopi, ERG (Elektroretinogramm), fargetest med oppmerksomhet for blå og

Слайд 9«Bone spicules», papilleatrofi
OFTALMOSKOPISK BILDE
«Bone spicules», papilleatrofi
OFTALMOSKOPISK BILDE
Слайд 10ME VED RP
ME VED RP
Слайд 11SENTRAL SF VED RP
SENTRAL SF VED RP

Слайд 12Ingen behandling visst seg effektiv
Vitamin A og E forsøk i høye doser
Ingen behandling visst seg effektiv
Vitamin A og E forsøk i høye doser

Слайд 13Prognose er generelt dårlig, sykdom progredierer livet ut
Kjønnsbundet former progredierer raskere
Prognose er generelt dårlig, sykdom progredierer livet ut
Kjønnsbundet former progredierer raskere

 Осмотр и диагностика болезней грызунов
Осмотр и диагностика болезней грызунов Сердечно-лёгочная реанимация
Сердечно-лёгочная реанимация Утопление
Утопление uporotaya_versia(1)
uporotaya_versia(1) Первая помощь при обмороках
Первая помощь при обмороках Российский Красный Крест
Российский Красный Крест Мифы о здоровом образе жизни
Мифы о здоровом образе жизни Профессия - охранять здоровье!
Профессия - охранять здоровье! Определение локального статуса хирургического больного
Определение локального статуса хирургического больного Вспомогательная репродуктивная технология
Вспомогательная репродуктивная технология Психология отношений в клинической практике
Психология отношений в клинической практике Дозирование препаратов инсулина в возрастном аспекте
Дозирование препаратов инсулина в возрастном аспекте Наследственные заболевания
Наследственные заболевания Гинекомастия и рак грудной железы
Гинекомастия и рак грудной железы Сестринский процесс при менингококковой инфекции и полиомиелите
Сестринский процесс при менингококковой инфекции и полиомиелите Презентация по диансеризации
Презентация по диансеризации Артериальное кровотечение. Признаки и оказания первой помощи
Артериальное кровотечение. Признаки и оказания первой помощи Мұнай өнімдерінің (бензол,толуол) теріге әсері
Мұнай өнімдерінің (бензол,толуол) теріге әсері Направление работы кадрового резерва: Здравоохранение
Направление работы кадрового резерва: Здравоохранение Основы латинского языка с медицинской терминологией
Основы латинского языка с медицинской терминологией Чистота – залог здоровья
Чистота – залог здоровья Спирт этиловый
Спирт этиловый ПРЕДМЕТ, ЗАДАЧИ И ОСНОВНЫЕ НАПРАВЛЕНИЯ КЛИНИЧЕСКОЙ ФАРМАКОЛОГИИ. ПОБОЧНЫЕ ЭФФЕКТЫ
ПРЕДМЕТ, ЗАДАЧИ И ОСНОВНЫЕ НАПРАВЛЕНИЯ КЛИНИЧЕСКОЙ ФАРМАКОЛОГИИ. ПОБОЧНЫЕ ЭФФЕКТЫ Медицинская аптечка
Медицинская аптечка Подготовка к зачёту
Подготовка к зачёту Центр диагностики и консультирования для детей, нуждающихся в психолого-педагогической и медико-социальной помощи
Центр диагностики и консультирования для детей, нуждающихся в психолого-педагогической и медико-социальной помощи Профилактика неинфекционных заболеваний и борьба с ними
Профилактика неинфекционных заболеваний и борьба с ними Profesionāla matu kopšanas līnija
Profesionāla matu kopšanas līnija