Влияние ядерной ДНК на механизмы возникновения дефектов в митохондриальной днк: миделеции (точечные мутации) мтднк,
- Влияние ядерной ДНК на механизмы возникновения дефектов в митохондриальной днк: миделеции (точечные мутации) мтднк,

Содержание
- 2. ЦЕЛЬ, АКТУАЛЬНОСТЬ, ЗАДАЧИ Актуальность: митохондриальные болезни составляют большую группу патологических состояний, связанных с генетически детерминированными нарушениями
- 3. Строение митохондриальной ДНК СТРОЕНИЕ МИТОХОНДРИАЛЬНОЙ ДНК Митохондрии произошли от древних симбиотических бактерий, которые жили внутри примитивных
- 4. Сравнение строения митохондриальной и ядерной ДНК ПРОИСХОЖДЕНИЕ И ДЕГРАДАЦИЯ МИТОХОНДРИАЛЬНОГО ГЕНОМА В ходе коэволюции оказалось, что
- 5. СРАВНЕНИЕ МИТОХОНДРИАЛЬНОЙ И ЯДЕРНОЙ ДНК
- 6. СРАВНЕНИЕ МИТОХОНДРИАЛЬНОЙ И ЯДЕРНОЙ ДНК
- 7. МИТОХОНДРИАЛЬНАЯ ДНК ПОД КОНТРОЛЕМ ЯДЕРНОЙ Известно, что синтез мтДНК находится под контролем ядерных генов. Мутации в
- 8. КЛАССИФИКАЦИЯ ПО ПАТОГЕНЕЗУ Митохондриальные заболевания являются результатом унаследованных и/или спонтанных мутаций в митохондриальной ДНК (мтДНК) и/или
- 9. КЛАССИФИКАЦИЯ ПО ЭТИОЛОГИИ Митохондриальные болезни, обусловленные генными мутациями ядерной ДНК Митохондриальные болезни, в основе которых лежат
- 10. ФЕНОТИПЫ ЯДЕРНО-КОДИРУЕМЫХ МИТОХОНДРИАЛЬНЫХ ЗАБОЛЕВАНИЙ
- 11. МИТОХОНДРИАЛЬНЫЕ СИНДРОМЫ, КОДИРУЕМЫЕ ЯДЕРНЫМИ ГЕНАМИ
- 12. ТОЧЕЧНЫЕ МУТАЦИИ МИТОХОНДРИАЛЬНОЙ ДНК В основе патогенеза синдрома MELAS лежат точечные мутации мтДНК, преимущественно генов транспортных
- 13. Атрофия зрительного нерва ДЕЛЕЦИИ МИТОХОНДРИАЛЬНОЙ ДНК, НАСЛЕДУЕМЫЕ ПО АУТОСОМНО-РЕЦЕССИВНОМУ ТИПУ Течение заболевания в виде митохондриальной энцефаломиопатии
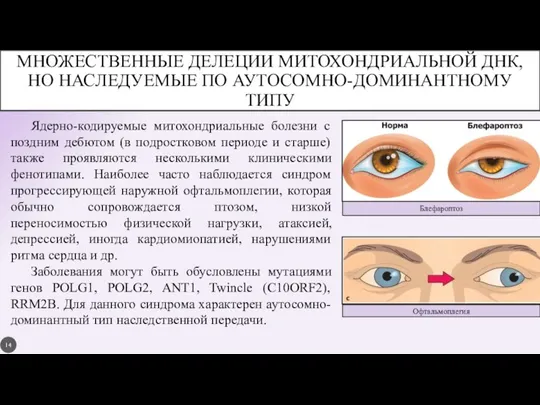
- 14. Офтальмоплегия Блефароптоз МНОЖЕСТВЕННЫЕ ДЕЛЕЦИИ МИТОХОНДРИАЛЬНОЙ ДНК, НО НАСЛЕДУЕМЫЕ ПО АУТОСОМНО-ДОМИНАНТНОМУ ТИПУ Ядерно-кодируемые митохондриальные болезни с поздним
- 15. ЗАКЛЮЧЕНИЕ Несмотря на небольшой размер, митохондриальный геном отвечает за правильное функционирование электростанций наших клеток. Этот кольцевой
- 16. СПИСОК ЛИТЕРАТУРЫ Harbauer, A.B., Zahedi, R.P., Sickmann, A., Pfanner, N., and Meisinger, C. (2014) The protein
- 18. Скачать презентацию
Слайд 2ЦЕЛЬ, АКТУАЛЬНОСТЬ, ЗАДАЧИ
Актуальность: митохондриальные болезни составляют большую группу патологических состояний, связанных с
ЦЕЛЬ, АКТУАЛЬНОСТЬ, ЗАДАЧИ
Актуальность: митохондриальные болезни составляют большую группу патологических состояний, связанных с

Слайд 3Строение митохондриальной ДНК
СТРОЕНИЕ МИТОХОНДРИАЛЬНОЙ ДНК
Митохондрии произошли от древних симбиотических бактерий, которые жили
Строение митохондриальной ДНК
СТРОЕНИЕ МИТОХОНДРИАЛЬНОЙ ДНК
Митохондрии произошли от древних симбиотических бактерий, которые жили

Слайд 4Сравнение строения митохондриальной и ядерной ДНК
ПРОИСХОЖДЕНИЕ И ДЕГРАДАЦИЯ МИТОХОНДРИАЛЬНОГО ГЕНОМА
В ходе коэволюции
Сравнение строения митохондриальной и ядерной ДНК
ПРОИСХОЖДЕНИЕ И ДЕГРАДАЦИЯ МИТОХОНДРИАЛЬНОГО ГЕНОМА
В ходе коэволюции

Слайд 5СРАВНЕНИЕ МИТОХОНДРИАЛЬНОЙ И ЯДЕРНОЙ ДНК
СРАВНЕНИЕ МИТОХОНДРИАЛЬНОЙ И ЯДЕРНОЙ ДНК

Слайд 6СРАВНЕНИЕ МИТОХОНДРИАЛЬНОЙ И ЯДЕРНОЙ ДНК
СРАВНЕНИЕ МИТОХОНДРИАЛЬНОЙ И ЯДЕРНОЙ ДНК

Слайд 7МИТОХОНДРИАЛЬНАЯ ДНК ПОД КОНТРОЛЕМ ЯДЕРНОЙ
Известно, что синтез мтДНК находится под контролем
МИТОХОНДРИАЛЬНАЯ ДНК ПОД КОНТРОЛЕМ ЯДЕРНОЙ
Известно, что синтез мтДНК находится под контролем

Слайд 8КЛАССИФИКАЦИЯ ПО ПАТОГЕНЕЗУ
Митохондриальные заболевания являются результатом унаследованных и/или спонтанных мутаций в митохондриальной
КЛАССИФИКАЦИЯ ПО ПАТОГЕНЕЗУ
Митохондриальные заболевания являются результатом унаследованных и/или спонтанных мутаций в митохондриальной

Слайд 9КЛАССИФИКАЦИЯ ПО ЭТИОЛОГИИ
Митохондриальные болезни, обусловленные генными мутациями ядерной ДНК
Митохондриальные болезни, в основе
КЛАССИФИКАЦИЯ ПО ЭТИОЛОГИИ
Митохондриальные болезни, обусловленные генными мутациями ядерной ДНК
Митохондриальные болезни, в основе

Слайд 10ФЕНОТИПЫ ЯДЕРНО-КОДИРУЕМЫХ МИТОХОНДРИАЛЬНЫХ ЗАБОЛЕВАНИЙ
ФЕНОТИПЫ ЯДЕРНО-КОДИРУЕМЫХ МИТОХОНДРИАЛЬНЫХ ЗАБОЛЕВАНИЙ

Слайд 11МИТОХОНДРИАЛЬНЫЕ СИНДРОМЫ, КОДИРУЕМЫЕ ЯДЕРНЫМИ ГЕНАМИ
МИТОХОНДРИАЛЬНЫЕ СИНДРОМЫ, КОДИРУЕМЫЕ ЯДЕРНЫМИ ГЕНАМИ

Слайд 12ТОЧЕЧНЫЕ МУТАЦИИ МИТОХОНДРИАЛЬНОЙ ДНК
В основе патогенеза синдрома MELAS лежат точечные мутации мтДНК,
ТОЧЕЧНЫЕ МУТАЦИИ МИТОХОНДРИАЛЬНОЙ ДНК
В основе патогенеза синдрома MELAS лежат точечные мутации мтДНК,

Слайд 13Атрофия зрительного нерва
ДЕЛЕЦИИ МИТОХОНДРИАЛЬНОЙ ДНК, НАСЛЕДУЕМЫЕ ПО АУТОСОМНО-РЕЦЕССИВНОМУ ТИПУ
Течение заболевания в виде
Атрофия зрительного нерва
ДЕЛЕЦИИ МИТОХОНДРИАЛЬНОЙ ДНК, НАСЛЕДУЕМЫЕ ПО АУТОСОМНО-РЕЦЕССИВНОМУ ТИПУ
Течение заболевания в виде

Слайд 14Офтальмоплегия
Блефароптоз
МНОЖЕСТВЕННЫЕ ДЕЛЕЦИИ МИТОХОНДРИАЛЬНОЙ ДНК, НО НАСЛЕДУЕМЫЕ ПО АУТОСОМНО-ДОМИНАНТНОМУ ТИПУ
Ядерно-кодируемые митохондриальные болезни с
Офтальмоплегия
Блефароптоз
МНОЖЕСТВЕННЫЕ ДЕЛЕЦИИ МИТОХОНДРИАЛЬНОЙ ДНК, НО НАСЛЕДУЕМЫЕ ПО АУТОСОМНО-ДОМИНАНТНОМУ ТИПУ
Ядерно-кодируемые митохондриальные болезни с
Слайд 15ЗАКЛЮЧЕНИЕ
Несмотря на небольшой размер, митохондриальный геном отвечает за правильное функционирование электростанций наших
ЗАКЛЮЧЕНИЕ
Несмотря на небольшой размер, митохондриальный геном отвечает за правильное функционирование электростанций наших

Слайд 16СПИСОК ЛИТЕРАТУРЫ
Harbauer, A.B., Zahedi, R.P., Sickmann, A., Pfanner, N., and Meisinger, C.
СПИСОК ЛИТЕРАТУРЫ
Harbauer, A.B., Zahedi, R.P., Sickmann, A., Pfanner, N., and Meisinger, C.

 Ревматическая лихорадка на Северном Кавказе: вчера, сегодня, завтра
Ревматическая лихорадка на Северном Кавказе: вчера, сегодня, завтра Метаболическое средство мельдоний
Метаболическое средство мельдоний Fast-food.империи McDonald’s. Польза или вред?
Fast-food.империи McDonald’s. Польза или вред? Фаст-фудтың химиялық құрамы мен адам ағзасына әсерін зерттеу
Фаст-фудтың химиялық құрамы мен адам ағзасына әсерін зерттеу АІИ кездесетін грамм оң бактериялар
АІИ кездесетін грамм оң бактериялар Предмет и задачи топографической анатомии и оперативной хирургии
Предмет и задачи топографической анатомии и оперативной хирургии Кровотечения
Кровотечения Нарушения звукопроизношения
Нарушения звукопроизношения Инструкции по возобновлению деятельности учреждения раннего образования за период ПОСТ-КОВИД 19
Инструкции по возобновлению деятельности учреждения раннего образования за период ПОСТ-КОВИД 19 Донорство крови. Акция Ты нужен
Донорство крови. Акция Ты нужен Возрастные особенности здоровья. Рекомендуемые пробы и индексы
Возрастные особенности здоровья. Рекомендуемые пробы и индексы Центр диагностики и консультирования для детей, нуждающихся в психолого-педагогической и медико-социальной помощи
Центр диагностики и консультирования для детей, нуждающихся в психолого-педагогической и медико-социальной помощи Лазерохирургическое лечение кондиломатоза у беременных
Лазерохирургическое лечение кондиломатоза у беременных Профилактика абортов в контексте решения демографической проблемы России
Профилактика абортов в контексте решения демографической проблемы России Анатомо-фізіологічні особливості ендокринної системи у дітей та семіотика уражень
Анатомо-фізіологічні особливості ендокринної системи у дітей та семіотика уражень To i owo o bakteriach lek.-dent. Agnieszka grucka
To i owo o bakteriach lek.-dent. Agnieszka grucka Положение пациента и доступ
Положение пациента и доступ Жуманов С Ноцицептикалық
Жуманов С Ноцицептикалық Повреждение. Некроз. Апоптоз
Повреждение. Некроз. Апоптоз Клинические аспекты применения современных антидепрессантов в психиатрической практике
Клинические аспекты применения современных антидепрессантов в психиатрической практике Гелиофобия – навязчивый страх, боязнь пребывания на солнце, избегание инсоляции
Гелиофобия – навязчивый страх, боязнь пребывания на солнце, избегание инсоляции Риккетсиозы. Определение
Риккетсиозы. Определение Метод исправления врожденных аномалий развития, опираясь на теорию морфогенеза Алана Тьюринга
Метод исправления врожденных аномалий развития, опираясь на теорию морфогенеза Алана Тьюринга Группы здоровья
Группы здоровья Психоорганические расстройства
Психоорганические расстройства Экзаменационный билет. Препараты
Экзаменационный билет. Препараты Мед.катастроф Лекция №10 травма груди
Мед.катастроф Лекция №10 травма груди Группы и категории инвалидности
Группы и категории инвалидности