- Врождённые наследственные заболевания

Содержание
- 2. Удельный вес врождённой и наследст-венной патологии в структуре заболе-ваемости и смертности новорождён-ных и детей раннего возраста
- 3. КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ Современная классификация наслед-ственных заболеваний по этиологичес-кому принципу основана на изучении родословных, клиническом обследова-нии,
- 4. ХРОМОСОМНЫЕ ЗАБОЛЕВАНИЯ Хромосомные заболевания включают в себя все состояния, характеризующиеся видимыми в световой микроскоп нарушениями структуры
- 5. Все эти состояния могут быть следствием разнообразных структурных перестроек хромосом (сбалансированные и несбалансированные транслокации, инверсии, делеции)
- 6. СИНДРОМ ДАУНА Наиболее часто диагностируемый хромосомный синдром. В результате скринирующих программ у беременных во втором или
- 7. СИНДРОМ ДЕЛЕЦИИ КОРОТКОГО ПЛЕЧА ХРОМОСОМЫ 5 Синдром 5р-, синдром Лежена, синдром «кошачьего крика» . Популяционная частота
- 8. СИНДРОМ ШЕРЕШЕВСКОГО–ТЕРНЕРА У большинства пациен-тов отсутствует одна Х-хромосома. При нали-чии мозаицизма с при-сутствующей Y-хромо-сомой (кариотип 45,
- 9. МОНОГЕННЫЕ ЗАБОЛЕВАНИЯ Моногенные заболевания характеризуются сходными признаками — определяются одним геном и наследуются согласно законам Менделя.
- 10. Фенилкетонурия также называется фенил-аланинемией, фенил-пировиноградной оли-гофренией. Заболева-ние относится к врож-денным нарушениям метаболизма , харак-теризуется повышени-ем уровня
- 11. МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ Митохондрии, как цитоплазматические ор-ганеллы, передаются от матери всему потомству (сперматозоиды содержат практически только ядерную
- 12. МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ Классическим примером митохондриальной патологии является наследственная оптическая нейропатия Лебера, однако современные исследования показывают вовлеченность
- 13. ДИСМОРФОЛОГИЯ Значительную часть патологии детского возраста занимают врождённые дефекты, то есть заболевания, обусловленные нарушением развития эмбриона
- 14. Возникают они наиболее часто, причиной их возникновения является в большинстве случаев взаимодействие генетических фак-торов и факторов
- 15. ВРОЖДЁННЫЕ ПОРОКИ РАЗВИТИЯ Приблизительно 2–3% новорождённых имеют серьезные ВПР. Эмбриологически такие дефекты классифицируются на три основных
- 16. ДЕФОРМАЦИИ Этот тип врождённых дефектов обнаруживается приблизительно у 1–2% новорождённых. Наиболее частыми дефектами являются косолапость, врождённый
- 17. ДИЗРУПЦИИ Точная частота дизрупций неизвестна, она выявляется у 1–2% новорождённых. Первым исследователем, описавшим данный вид патологии
- 18. ЧАСТНАЯ СИНДРОМОЛОГИЯ C точки зрения практической неонатологии все синдромальные формы патологии новорождённых можно разделить на три
- 19. СИНДРОМ БЕКВИТА–ВИДЕМАННА Диагноз этого заболевания необходимо рас-сматривать у детей с эмбриональной или пупочной грыжей, макроглоссией, неонатальной
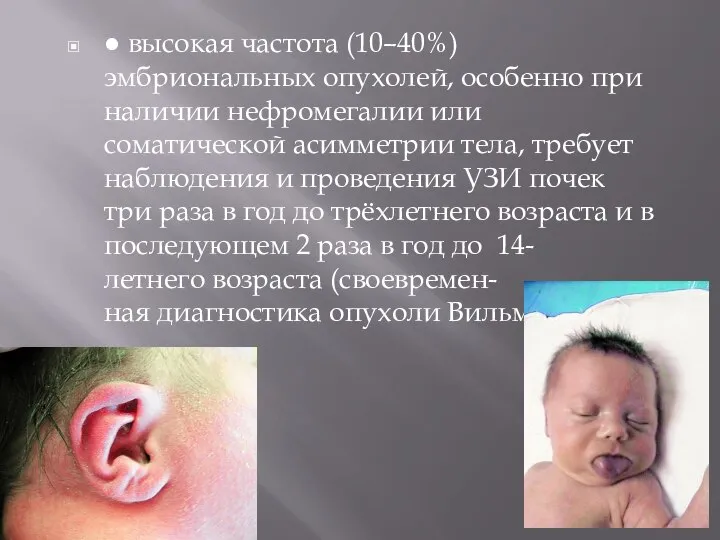
- 20. ● высокая частота (10–40%) эмбриональных опухолей, особенно при наличии нефромегалии или соматической асимметрии тела, требует наблюдения
- 21. СИНДРОМ НУНАН Наследственное аутосомно-доминантное заболевание. В 50% случаев возможна молекулярно генетическая верификация мутаций гена PTPN11. У
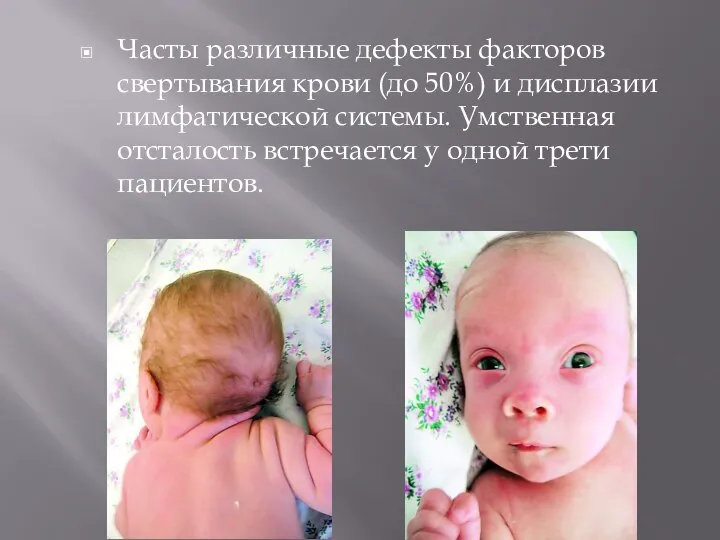
- 22. Часты различные дефекты факторов свертывания крови (до 50%) и дисплазии лимфатической системы. Умственная отсталость встречается у
- 24. Скачать презентацию
Слайд 2Удельный вес врождённой и наследст-венной патологии в структуре заболе-ваемости и смертности новорождён-ных
Удельный вес врождённой и наследст-венной патологии в структуре заболе-ваемости и смертности новорождён-ных

Слайд 3
КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Современная классификация наслед-ственных заболеваний по этиологичес-кому принципу основана на изучении
КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Современная классификация наслед-ственных заболеваний по этиологичес-кому принципу основана на изучении
Слайд 4ХРОМОСОМНЫЕ ЗАБОЛЕВАНИЯ
Хромосомные заболевания включают в себя все состояния, характеризующиеся видимыми в световой
ХРОМОСОМНЫЕ ЗАБОЛЕВАНИЯ
Хромосомные заболевания включают в себя все состояния, характеризующиеся видимыми в световой

Слайд 5Все эти состояния могут быть следствием разнообразных структурных перестроек хромосом (сбалансированные и
Все эти состояния могут быть следствием разнообразных структурных перестроек хромосом (сбалансированные и

Слайд 6СИНДРОМ ДАУНА
Наиболее часто диагностируемый хромосомный синдром. В результате скринирующих программ у беременных
СИНДРОМ ДАУНА
Наиболее часто диагностируемый хромосомный синдром. В результате скринирующих программ у беременных

Слайд 7
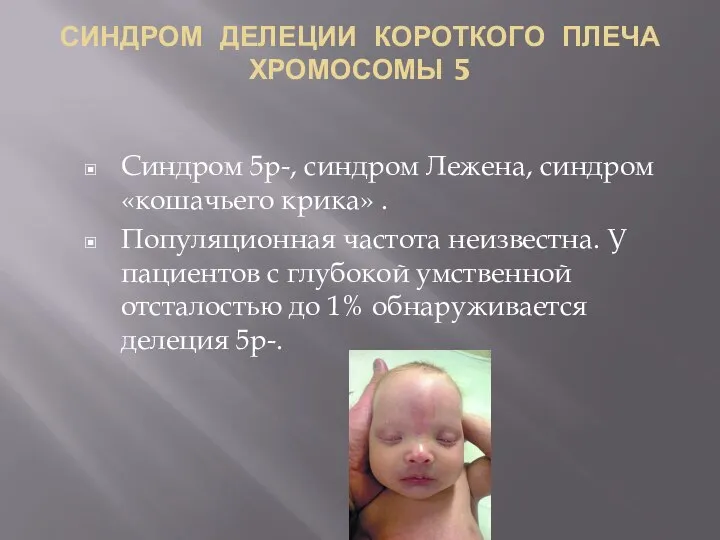
СИНДРОМ ДЕЛЕЦИИ КОРОТКОГО ПЛЕЧА ХРОМОСОМЫ 5
Синдром 5р-, синдром Лежена, синдром «кошачьего
СИНДРОМ ДЕЛЕЦИИ КОРОТКОГО ПЛЕЧА ХРОМОСОМЫ 5
Синдром 5р-, синдром Лежена, синдром «кошачьего
Слайд 8
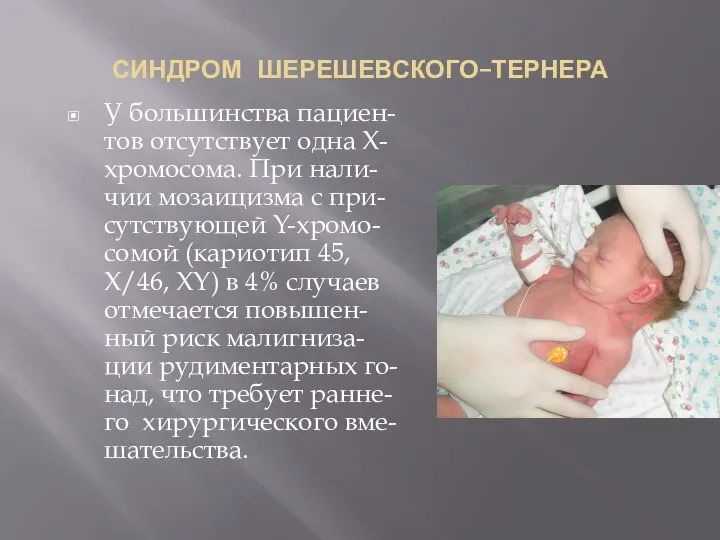
СИНДРОМ ШЕРЕШЕВСКОГО–ТЕРНЕРА
У большинства пациен-тов отсутствует одна Х-хромосома. При нали-чии мозаицизма с при-сутствующей
СИНДРОМ ШЕРЕШЕВСКОГО–ТЕРНЕРА
У большинства пациен-тов отсутствует одна Х-хромосома. При нали-чии мозаицизма с при-сутствующей
Слайд 9МОНОГЕННЫЕ ЗАБОЛЕВАНИЯ
Моногенные заболевания характеризуются сходными признаками — определяются одним геном и наследуются согласно
МОНОГЕННЫЕ ЗАБОЛЕВАНИЯ
Моногенные заболевания характеризуются сходными признаками — определяются одним геном и наследуются согласно

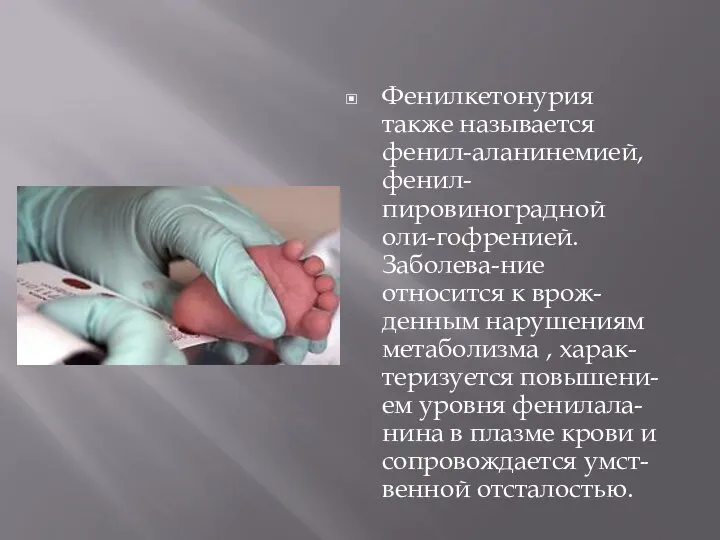
Слайд 10Фенилкетонурия также называется фенил-аланинемией, фенил-пировиноградной оли-гофренией. Заболева-ние относится к врож-денным нарушениям метаболизма
Фенилкетонурия также называется фенил-аланинемией, фенил-пировиноградной оли-гофренией. Заболева-ние относится к врож-денным нарушениям метаболизма
Слайд 11МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
Митохондрии, как цитоплазматические ор-ганеллы, передаются от матери всему потомству (сперматозоиды содержат
МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
Митохондрии, как цитоплазматические ор-ганеллы, передаются от матери всему потомству (сперматозоиды содержат

Слайд 12МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
Классическим примером митохондриальной патологии является наследственная оптическая нейропатия Лебера, однако современные
МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
Классическим примером митохондриальной патологии является наследственная оптическая нейропатия Лебера, однако современные

Слайд 13ДИСМОРФОЛОГИЯ
Значительную часть патологии детского возраста занимают врождённые дефекты, то есть заболевания, обусловленные
ДИСМОРФОЛОГИЯ
Значительную часть патологии детского возраста занимают врождённые дефекты, то есть заболевания, обусловленные

Слайд 14Возникают они наиболее часто, причиной их возникновения является в большинстве случаев взаимодействие
Возникают они наиболее часто, причиной их возникновения является в большинстве случаев взаимодействие

Слайд 15ВРОЖДЁННЫЕ ПОРОКИ РАЗВИТИЯ
Приблизительно 2–3% новорождённых имеют серьезные ВПР. Эмбриологически такие дефекты классифицируются
ВРОЖДЁННЫЕ ПОРОКИ РАЗВИТИЯ
Приблизительно 2–3% новорождённых имеют серьезные ВПР. Эмбриологически такие дефекты классифицируются

Слайд 16ДЕФОРМАЦИИ
Этот тип врождённых дефектов обнаруживается приблизительно у 1–2% новорождённых. Наиболее частыми дефектами
ДЕФОРМАЦИИ
Этот тип врождённых дефектов обнаруживается приблизительно у 1–2% новорождённых. Наиболее частыми дефектами

Слайд 17ДИЗРУПЦИИ
Точная частота дизрупций неизвестна, она выявляется у 1–2% новорождённых. Первым исследователем, описавшим
ДИЗРУПЦИИ
Точная частота дизрупций неизвестна, она выявляется у 1–2% новорождённых. Первым исследователем, описавшим

Слайд 18ЧАСТНАЯ СИНДРОМОЛОГИЯ
C точки зрения практической неонатологии все синдромальные формы патологии новорождённых можно
ЧАСТНАЯ СИНДРОМОЛОГИЯ
C точки зрения практической неонатологии все синдромальные формы патологии новорождённых можно

Слайд 19СИНДРОМ БЕКВИТА–ВИДЕМАННА
Диагноз этого заболевания необходимо рас-сматривать у детей с эмбриональной или пупочной
СИНДРОМ БЕКВИТА–ВИДЕМАННА
Диагноз этого заболевания необходимо рас-сматривать у детей с эмбриональной или пупочной

Слайд 20● высокая частота (10–40%) эмбриональных опухолей, особенно при наличии нефромегалии или соматической асимметрии
● высокая частота (10–40%) эмбриональных опухолей, особенно при наличии нефромегалии или соматической асимметрии
Слайд 21СИНДРОМ НУНАН
Наследственное аутосомно-доминантное заболевание. В 50% случаев возможна молекулярно генетическая верификация мутаций
СИНДРОМ НУНАН
Наследственное аутосомно-доминантное заболевание. В 50% случаев возможна молекулярно генетическая верификация мутаций

Слайд 22Часты различные дефекты факторов свертывания крови (до 50%) и дисплазии лимфатической системы.
Часты различные дефекты факторов свертывания крови (до 50%) и дисплазии лимфатической системы.
 Тактика участкового терапевта при острой и хронической диарее: факторы риска, диагностический алгоритм
Тактика участкового терапевта при острой и хронической диарее: факторы риска, диагностический алгоритм Психоактивные вещества: мифы и реалии
Психоактивные вещества: мифы и реалии Национальный медицинский исследовательский центр профилактической медицины
Национальный медицинский исследовательский центр профилактической медицины Ребенок 0-3 мес
Ребенок 0-3 мес Параэзофагеальные грыжи пищеводного отверстия диафрагмы
Параэзофагеальные грыжи пищеводного отверстия диафрагмы Российское общество профилактики неинфекционных заболеваний
Российское общество профилактики неинфекционных заболеваний Варикозное расширение вен
Варикозное расширение вен Дифтерия. История
Дифтерия. История Внутрикожные инъекции
Внутрикожные инъекции Трансплантационный иммунитет
Трансплантационный иммунитет Изучение основных положений организации рационального питания и освоение методов его гигиенической оценки
Изучение основных положений организации рационального питания и освоение методов его гигиенической оценки Иммуно- микробиологические исследования
Иммуно- микробиологические исследования Постменопаузальный период
Постменопаузальный период Центр по лечению ХСН, маршрутизация пациентов с ХСН
Центр по лечению ХСН, маршрутизация пациентов с ХСН Первая медицинская помощь при кровотечениях. Тема 18
Первая медицинская помощь при кровотечениях. Тема 18 Алалия. методы и приёмы коррекции
Алалия. методы и приёмы коррекции Прикладная анатомия брюшной стенки. Техника лапароскопии
Прикладная анатомия брюшной стенки. Техника лапароскопии МДК02.03 Л21 № 3 ч.2 акушеркам А21 Основы трансфузиологии
МДК02.03 Л21 № 3 ч.2 акушеркам А21 Основы трансфузиологии Краткосрочные курсы Сам себе спасатель
Краткосрочные курсы Сам себе спасатель Транзиторные состояния неонатального периода
Транзиторные состояния неонатального периода Беда по имени СПИД
Беда по имени СПИД Заболевания глаз, удаление инородных тел из глаза, уха, носа
Заболевания глаз, удаление инородных тел из глаза, уха, носа Виды бородавок. Предрасполагающие факторы
Виды бородавок. Предрасполагающие факторы Проект на тему: мы за здоровое питание
Проект на тему: мы за здоровое питание Пролежни
Пролежни Предоперационная подготовка хирурга
Предоперационная подготовка хирурга Статистика. Роскошь или необходимость?
Статистика. Роскошь или необходимость? Руброфития гладкой кожи
Руброфития гладкой кожи