Адиабатическое приближение, потенциальные поверхности молекулярных систем, неадиабатические переходы
- Адиабатическое приближение, потенциальные поверхности молекулярных систем, неадиабатические переходы

Содержание
- 2. УРАВНЕНИЕ ШРЕДИНГЕРА МОЛЕКУЛЯРНОЙ СИСТЕМЫ В химии широко используются такие понятия, как геометрическая структура молекулы, ее колебания
- 3. АДИАБАТИЧЕСКОЕ ПРИБИЖЕНИЕ ФИЗИЧЕСКОЕ ОБОСНОВАНИЕ И ОСНОВНЫЕ ВЫВОДЫ Адиабатическое приближение (приближение Борна-Оппенгеймера) me/Mядра ~ 10-4 ⇓ Электроны
- 4. АДИАБАТИЧЕСКОЕ ПРИБИЖЕНИЕ МАТЕМАТИЧЕСКАЯ ФОРМУЛИРОВКА Полный базисный набор из собственных функций электронного гамильтониана при фиксированных конфигурациях ядер
- 5. ПОТЕНЦИАЛЬНЫЕ КРИВЫЕ ДВУХАТОМНОЙ МОЛЕКУЛЫ Экспериментальные притягивающие потенциальные кривые молекулы SO Малые межъядерные расстояния R: U (R)
- 6. КАК ВЗАИМОДЕЙСТВУЕТ АТОМ F С АТОМОМ Kr? Они отталкиваются, т.к. Kr благородный газ. Ответ неправильный, т.к.
- 7. ДИССОЦИАЦИЯ KCl НА K+ И Cl- - НЕАДИАБАТИЧЕСКИЙ ПРОЦЕСС Вероятности адиабатической и неадиабатической диссоциации (нестационарных процессов),
- 8. ТУШЕНИЕ ИЗЛУЧАЮЩЕГО МОЛЕКУЛЯРНОГО СОСТОЯНИЯ ПРИ СТОЛКНОВЕНИИ Расщепление индуцируется взаимодействием Na2 с атомом инертного газа Скорость тушения
- 9. ЭЛЕКТРОННЫЕ ПОТЕНЦИАЛЬНЫЕ ПОВЕРХНОСТИ, НА КОТОРЫХ ПРОТЕКАЮТ ХИМИЧЕСКИЕ РЕАКЦИИ Lr - путь реакции qr – координата реакции
- 10. КАК ВОЗНИКАЕТ ПОТЕНЦИАЛЬНЫЙ БАРЬЕР ? Проблема классической химии – почему энергия активации существенно меньше энергий связи
- 11. ПРОФИЛИ ПУТИ АДИАБАТИЧЕСКИХ ХИМИЧЕСКИХ РЕАКЦИЙ РАЗЛИЧНЫХ ТИПОВ Реакция между двумя радикалами с образованием двух радикалов Реакция
- 13. Скачать презентацию
Слайд 2УРАВНЕНИЕ ШРЕДИНГЕРА МОЛЕКУЛЯРНОЙ СИСТЕМЫ
В химии широко используются такие понятия, как геометрическая структура
УРАВНЕНИЕ ШРЕДИНГЕРА МОЛЕКУЛЯРНОЙ СИСТЕМЫ
В химии широко используются такие понятия, как геометрическая структура

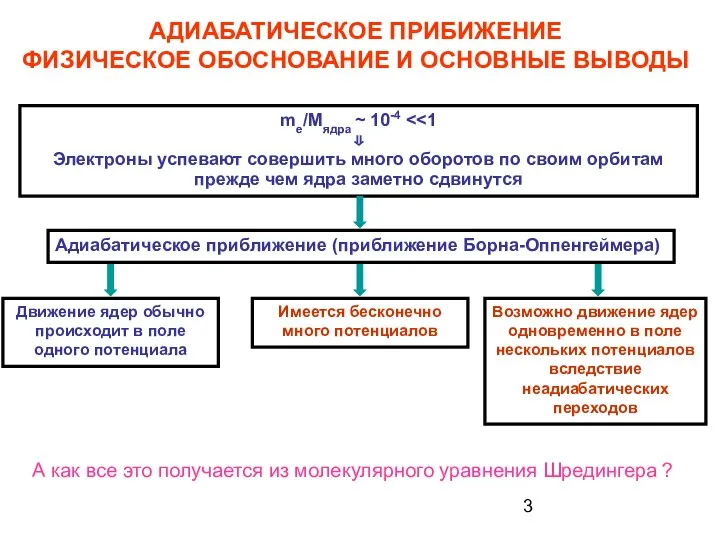
Слайд 3АДИАБАТИЧЕСКОЕ ПРИБИЖЕНИЕ
ФИЗИЧЕСКОЕ ОБОСНОВАНИЕ И ОСНОВНЫЕ ВЫВОДЫ
Адиабатическое приближение (приближение Борна-Оппенгеймера)
me/Mядра ~ 10-4 <<1
АДИАБАТИЧЕСКОЕ ПРИБИЖЕНИЕ
ФИЗИЧЕСКОЕ ОБОСНОВАНИЕ И ОСНОВНЫЕ ВЫВОДЫ
Адиабатическое приближение (приближение Борна-Оппенгеймера)
me/Mядра ~ 10-4 <<1
Слайд 4АДИАБАТИЧЕСКОЕ ПРИБИЖЕНИЕ
МАТЕМАТИЧЕСКАЯ ФОРМУЛИРОВКА
Полный базисный набор из собственных функций электронного гамильтониана при
АДИАБАТИЧЕСКОЕ ПРИБИЖЕНИЕ
МАТЕМАТИЧЕСКАЯ ФОРМУЛИРОВКА
Полный базисный набор из собственных функций электронного гамильтониана при

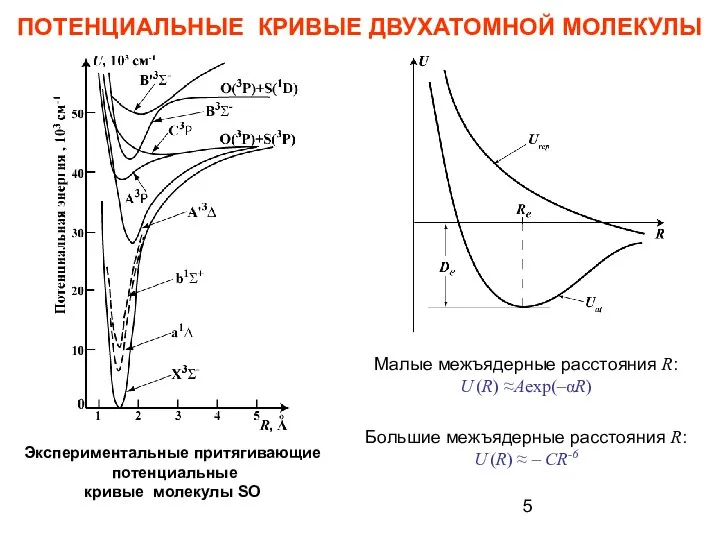
Слайд 5ПОТЕНЦИАЛЬНЫЕ КРИВЫЕ ДВУХАТОМНОЙ МОЛЕКУЛЫ
Экспериментальные притягивающие
потенциальные
кривые молекулы SO
Малые межъядерные расстояния R:
ПОТЕНЦИАЛЬНЫЕ КРИВЫЕ ДВУХАТОМНОЙ МОЛЕКУЛЫ
Экспериментальные притягивающие
потенциальные
кривые молекулы SO
Малые межъядерные расстояния R:
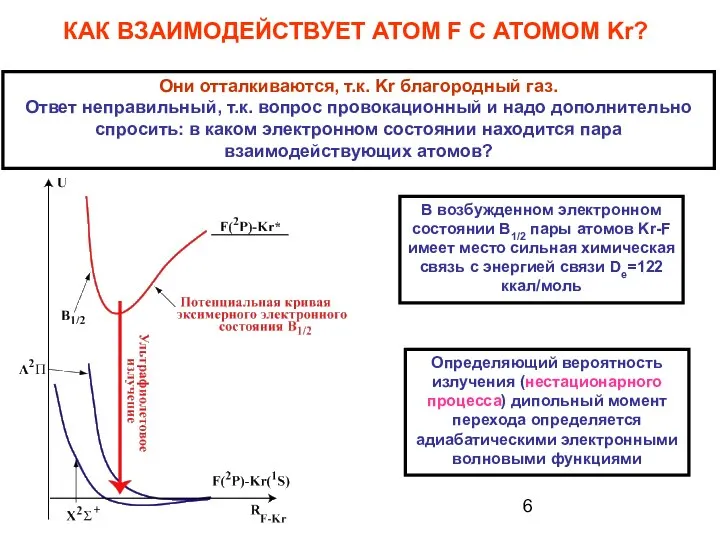
Слайд 6КАК ВЗАИМОДЕЙСТВУЕТ АТОМ F С АТОМОМ Kr?
Они отталкиваются, т.к. Kr благородный газ.
Ответ
КАК ВЗАИМОДЕЙСТВУЕТ АТОМ F С АТОМОМ Kr?
Они отталкиваются, т.к. Kr благородный газ.
Ответ
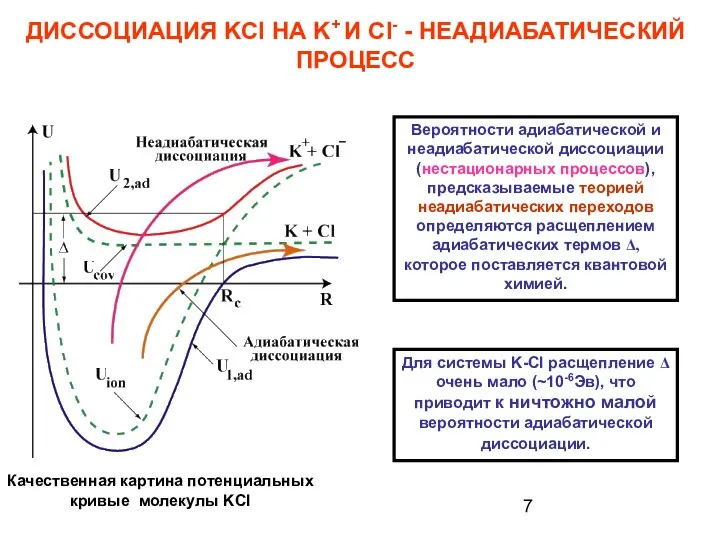
Слайд 7ДИССОЦИАЦИЯ KCl НА K+ И Cl- - НЕАДИАБАТИЧЕСКИЙ ПРОЦЕСС
Вероятности адиабатической и
ДИССОЦИАЦИЯ KCl НА K+ И Cl- - НЕАДИАБАТИЧЕСКИЙ ПРОЦЕСС
Вероятности адиабатической и
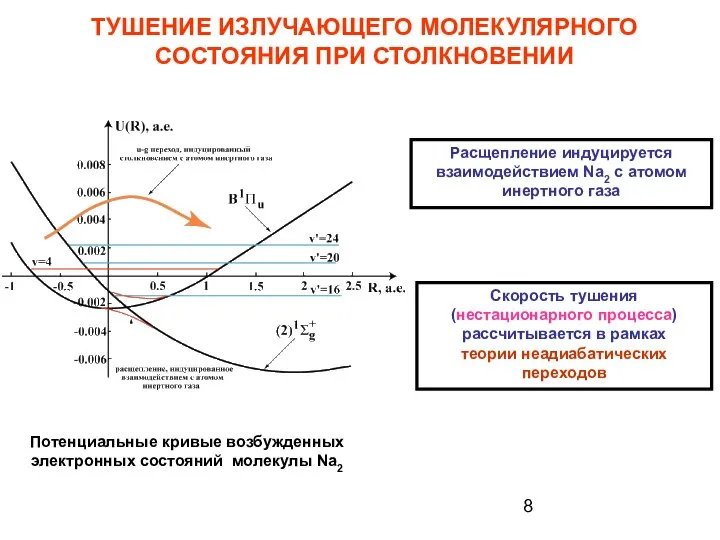
Слайд 8ТУШЕНИЕ ИЗЛУЧАЮЩЕГО МОЛЕКУЛЯРНОГО
СОСТОЯНИЯ ПРИ СТОЛКНОВЕНИИ
Расщепление индуцируется взаимодействием Na2 с атомом инертного
ТУШЕНИЕ ИЗЛУЧАЮЩЕГО МОЛЕКУЛЯРНОГО
СОСТОЯНИЯ ПРИ СТОЛКНОВЕНИИ
Расщепление индуцируется взаимодействием Na2 с атомом инертного
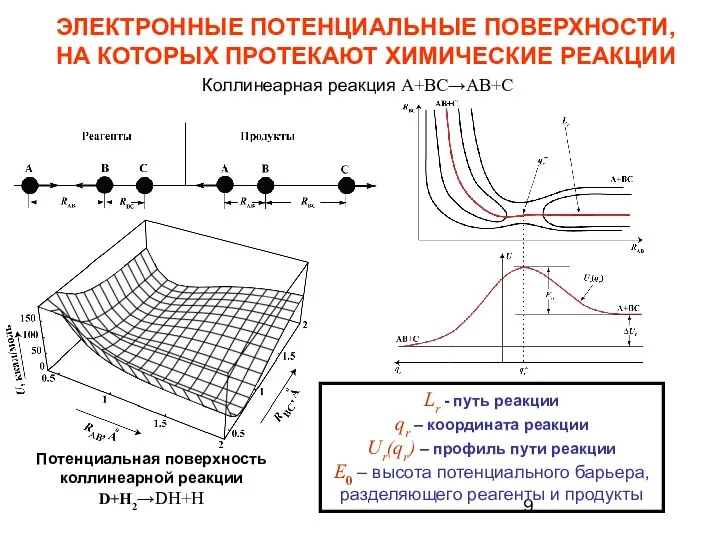
Слайд 9ЭЛЕКТРОННЫЕ ПОТЕНЦИАЛЬНЫЕ ПОВЕРХНОСТИ,
НА КОТОРЫХ ПРОТЕКАЮТ ХИМИЧЕСКИЕ РЕАКЦИИ
Lr - путь реакции
qr – координата
ЭЛЕКТРОННЫЕ ПОТЕНЦИАЛЬНЫЕ ПОВЕРХНОСТИ,
НА КОТОРЫХ ПРОТЕКАЮТ ХИМИЧЕСКИЕ РЕАКЦИИ
Lr - путь реакции
qr – координата
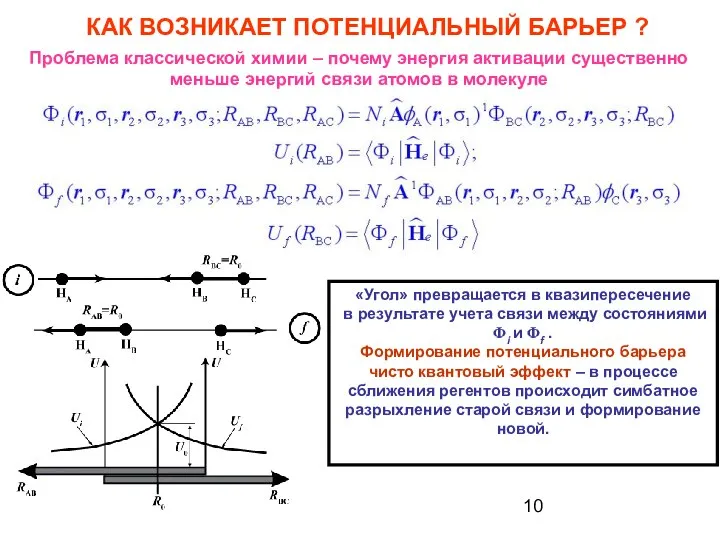
Слайд 10КАК ВОЗНИКАЕТ ПОТЕНЦИАЛЬНЫЙ БАРЬЕР ?
Проблема классической химии – почему энергия активации существенно
меньше
КАК ВОЗНИКАЕТ ПОТЕНЦИАЛЬНЫЙ БАРЬЕР ?
Проблема классической химии – почему энергия активации существенно
меньше
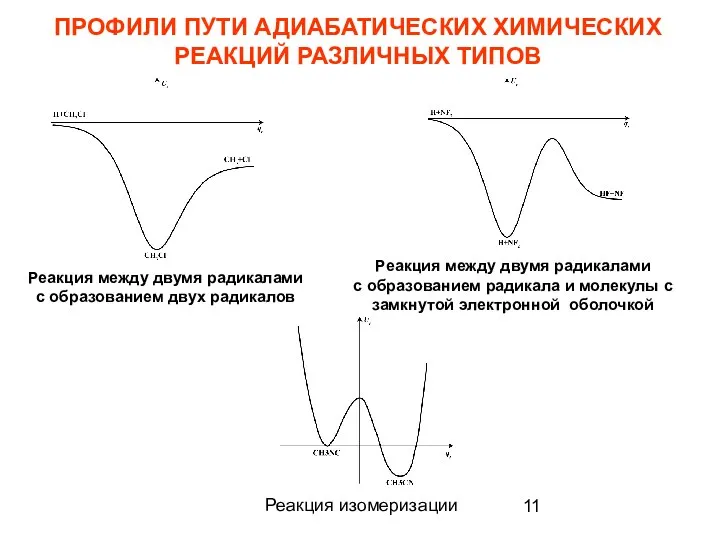
Слайд 11ПРОФИЛИ ПУТИ АДИАБАТИЧЕСКИХ ХИМИЧЕСКИХ
РЕАКЦИЙ РАЗЛИЧНЫХ ТИПОВ
Реакция между двумя радикалами
с образованием двух
ПРОФИЛИ ПУТИ АДИАБАТИЧЕСКИХ ХИМИЧЕСКИХ
РЕАКЦИЙ РАЗЛИЧНЫХ ТИПОВ
Реакция между двумя радикалами
с образованием двух
 Природные и синтетические красители
Природные и синтетические красители Типы химических связей 8 класс
Типы химических связей 8 класс Металлы в природе. Общие способы их получения
Металлы в природе. Общие способы их получения Презентация на тему: Подгруппа азота
Презентация на тему: Подгруппа азота Нуклеинови киселини
Нуклеинови киселини Спирты
Спирты Количество вещества, число Авогадро, молярная масса, молярный объём, уравнение связи
Количество вещества, число Авогадро, молярная масса, молярный объём, уравнение связи Влияние бытовой химии на здоровье человека
Влияние бытовой химии на здоровье человека alkany
alkany bc95487a97b0472591b20aef8d569081
bc95487a97b0472591b20aef8d569081 каталитический крекинг
каталитический крекинг Обсидиан
Обсидиан _Периодический закон. Распределение электронов по энергетическим уровням и подуровням (1)
_Периодический закон. Распределение электронов по энергетическим уровням и подуровням (1) Основные классы неорганических соединений
Основные классы неорганических соединений Строение, свойства, биологическая роль дезоксисахаров и аминосахаров
Строение, свойства, биологическая роль дезоксисахаров и аминосахаров Работа ученика 9 «в» класса МОУ «СОШ №59» Попова Михаила Руководитель Самсонова Г.М.
Работа ученика 9 «в» класса МОУ «СОШ №59» Попова Михаила Руководитель Самсонова Г.М. Фосфолипиды. Глицерофосфолипиды. Сфингофосфолипиды. Гликолипиды
Фосфолипиды. Глицерофосфолипиды. Сфингофосфолипиды. Гликолипиды Искусственные полимеры
Искусственные полимеры Ионная химическая связь
Ионная химическая связь Турнир знатоков химии. Химическая лихорадка
Турнир знатоков химии. Химическая лихорадка Презентация по Химии "Фосфор - элемент жизни и мысли"
Презентация по Химии "Фосфор - элемент жизни и мысли"  Презентация на тему Жиры (9 класс)
Презентация на тему Жиры (9 класс)  АРЕНЫ
АРЕНЫ Двовимірний ямр. Основні принципи
Двовимірний ямр. Основні принципи Виды металлов, сплавы металлов, применение в промышленности и в быту
Виды металлов, сплавы металлов, применение в промышленности и в быту Количество вещества - моль
Количество вещества - моль Классификация неорганических веществ
Классификация неорганических веществ Проблемное обучение на уроках химии
Проблемное обучение на уроках химии