- Біоеквівалентність як один з видів клінічних випробувань

Содержание
- 2. У клінічній практиці вже давно відомо, що препарати, що мають одні і ті ж активні речовини,
- 3. Біологічна доступність Біодоступність - це швидкість і ступінь, з яким активна субстанція накопичується в місці її
- 4. Процес вивільнення лікарської речовини часто є чинником, лімітуючим швидкість всмоктування в тих випадках, коли препарат призначають
- 5. Препарат-генерик В даний час дослідження біодоступності/біоеквівалентності лікарських засобів вважається основним видом медико-біологічного контролю якості препаратів, особливо
- 6. Біоеквівалентність – порівняльна біодоступність двох препаратів Вимога ефективності і безпеки генеричних лікарських препаратів, вироблюваних різними фірмами,
- 7. Біологічна нееквівалентність Терміном біологічна нееквівалентність лікарських препаратів позначається невідповідність одних і тих же препаратів, що випускаються
- 8. Значення досліджень по біоеквівалентності Якщо неможливо встановити причину зміни реакції організму на лікарський препарат, це може
- 9. Регламент проведення досліджень по біоеквівалентності У зв'язку з цим, в 1991 р. в Канаді і в
- 10. Особливості препаратів-генериків Генерічеськоє лікарський засіб містить активна лікарська речовина (активну субстанцію) ідентичне активній речовині оригінального (патентованого)
- 11. Генеричний лікарський препарат може розглядатися як взаємозамінний оригінального препарату тільки в тому випадку, якщо він відповідає
- 12. Вимоги до виробництва препаратів-генериків Для генеричних препаратів необхідно: при виробництві - дотримання вимог належної виробничої практики
- 13. Визначення біоеквівалентності Біоеквівалентність - два лікарські препарати вважаються біоеквівалентними, якщо вони фармацевтично еквівалентні і їх біодоступність
- 14. Особливості при проведенні дослідження по біоеквівалентності Необхідно підкреслити, що вивчення біоеквівалентності - це клінічні випробування, де
- 15. Вимоги до випробовуваних Контингент досліджуваних для вивчення біоеквівалентності повинен бути якомога одноріднішим, тому дослідження повинне проводитися
- 16. Можливість пацієнтів (здорових добровольців) брати участь в дослідженні повинна підтверджуватися даними, отриманими при використанні стандартних лабораторних
- 17. Кількість випробовуваних для проведення дослідження Мінімальне число випробовуваних, таких, що залучаються до досліджень біоеквівалентності, складає 12
- 18. Особливості режиму для випробовуваних при проведенні дослідження З метою мінімізації відмінностей, що виникають під впливом чинників,
- 19. Особливості дизайну випробування Особливістю дизайну таких досліджень є те, що кожний з випробовуваних отримує як стандартний
- 20. Фармкокінетичні параметри, які визначаються для оцінки біодоступності і біоеквівалентності При вивченні біодоступності лікарських препаратів найбільш важливими
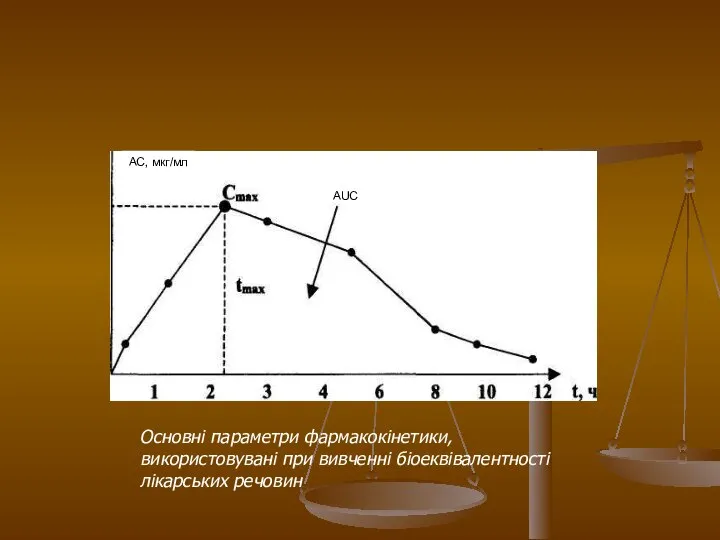
- 21. АС, мкг/мл Основні параметри фармакокінетики, використовувані при вивченні біоеквівалентності лікарських речовин
- 22. При вивченні біодоступності методом введення одноразової дози площа під кривою «концентрація-час» визначається після одноразового введення лікарського
- 23. Час відбору проб для дослідження Час узяття проб крові у випробовуваних необхідно планувати так, щоб забезпечити
- 24. Значення показника максимальної концентрації речовини можна пояснити за допомогою наступного прикладу. На малюнку 2 представлені фармакокінетичні
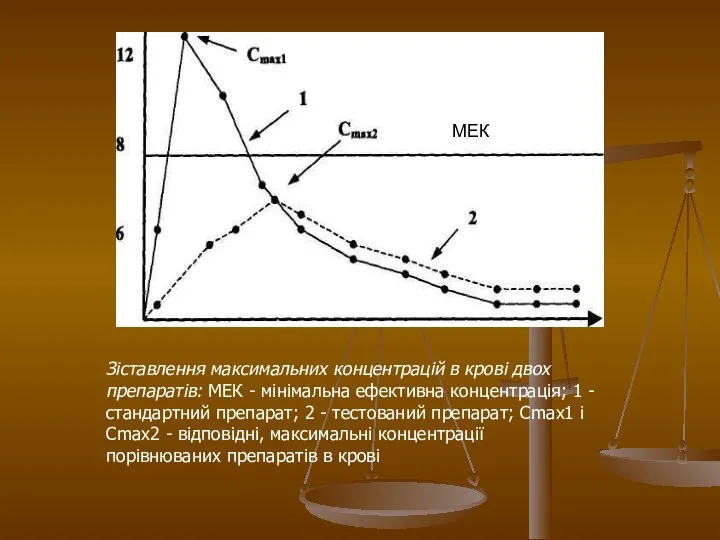
- 25. Зіставлення максимальних концентрацій в крові двох препаратів: МЕК - мінімальна ефективна концентрація; 1 - стандартний препарат;
- 26. Крива 1 характеризує концентрацію в крові стандартного препарату, а крива 2 - тестованого. Горизонтальною лінією відмічена
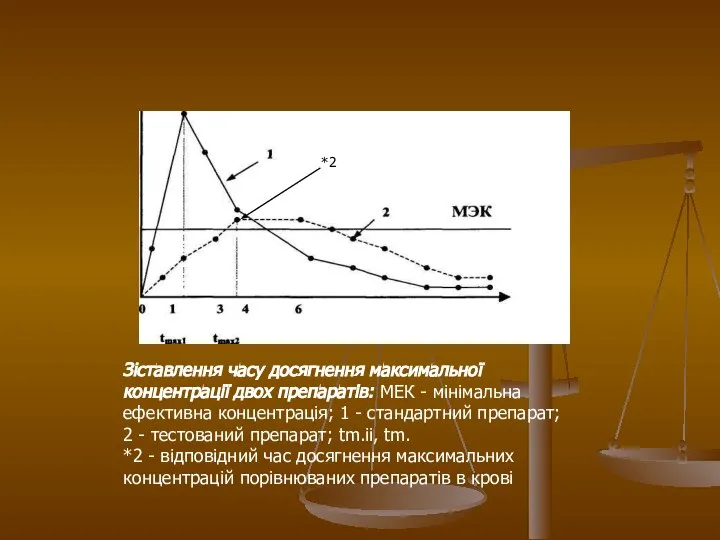
- 27. На малюнку 3 показано, що максимальна концентрація стандартного препарату (крива 1) досягається через 1 годину, а
- 28. Зіставлення часу досягнення максимальної концентрації двох препаратів: МЕК - мінімальна ефективна концентрація; 1 - стандартний препарат;
- 29. На малюнку 4 представлений інший приклад співвідношення кривих, що відображають кінетику двох порівнюваних препаратів. Площа під
- 30. Зіставлення площ під фармакокинетичними кривими двох препаратів: 1 - стандартний препарат; 2 - тестований препарат; AUC1,
- 31. Таким чином, два препарати вважаються біоеквівалентними, якщо вони мають схожі фармакокінетичні показники. За регламентом ВОЗ (1994,
- 32. Звіт про проведене випробування Всі етапи проведення дослідження повинні ретельно фіксуватися, аналізуватися, описуватися, відповідно до заздалегідь
- 34. Скачать презентацию

Слайд 3Біологічна доступність
Біодоступність - це швидкість і ступінь, з яким активна субстанція накопичується
Біологічна доступність
Біодоступність - це швидкість і ступінь, з яким активна субстанція накопичується

Слайд 4Процес вивільнення лікарської речовини часто є чинником, лімітуючим швидкість всмоктування в тих
Процес вивільнення лікарської речовини часто є чинником, лімітуючим швидкість всмоктування в тих

Слайд 5Препарат-генерик
В даний час дослідження біодоступності/біоеквівалентності лікарських засобів вважається основним видом медико-біологічного контролю
Препарат-генерик
В даний час дослідження біодоступності/біоеквівалентності лікарських засобів вважається основним видом медико-біологічного контролю

Слайд 6Біоеквівалентність – порівняльна біодоступність двох препаратів
Вимога ефективності і безпеки генеричних лікарських препаратів,
Біоеквівалентність – порівняльна біодоступність двох препаратів
Вимога ефективності і безпеки генеричних лікарських препаратів,

Слайд 7Біологічна нееквівалентність
Терміном біологічна нееквівалентність лікарських препаратів позначається невідповідність одних і тих же
Біологічна нееквівалентність
Терміном біологічна нееквівалентність лікарських препаратів позначається невідповідність одних і тих же

Слайд 8Значення досліджень по біоеквівалентності
Якщо неможливо встановити причину зміни реакції організму на лікарський
Значення досліджень по біоеквівалентності
Якщо неможливо встановити причину зміни реакції організму на лікарський

Слайд 9Регламент проведення досліджень по біоеквівалентності
У зв'язку з цим, в 1991 р. в
Регламент проведення досліджень по біоеквівалентності
У зв'язку з цим, в 1991 р. в

Слайд 10Особливості препаратів-генериків
Генерічеськоє лікарський засіб містить активна лікарська речовина (активну субстанцію) ідентичне активній
Особливості препаратів-генериків
Генерічеськоє лікарський засіб містить активна лікарська речовина (активну субстанцію) ідентичне активній

Слайд 11Генеричний лікарський препарат може розглядатися як взаємозамінний оригінального препарату тільки в тому
Генеричний лікарський препарат може розглядатися як взаємозамінний оригінального препарату тільки в тому

Слайд 12Вимоги до виробництва препаратів-генериків
Для генеричних препаратів необхідно:
при виробництві - дотримання вимог належної
Вимоги до виробництва препаратів-генериків
Для генеричних препаратів необхідно:
при виробництві - дотримання вимог належної

Слайд 13Визначення біоеквівалентності
Біоеквівалентність - два лікарські препарати вважаються біоеквівалентними, якщо вони фармацевтично еквівалентні
Визначення біоеквівалентності
Біоеквівалентність - два лікарські препарати вважаються біоеквівалентними, якщо вони фармацевтично еквівалентні

Слайд 14Особливості при проведенні дослідження по біоеквівалентності
Необхідно підкреслити, що вивчення біоеквівалентності - це
Особливості при проведенні дослідження по біоеквівалентності
Необхідно підкреслити, що вивчення біоеквівалентності - це

Слайд 15Вимоги до випробовуваних
Контингент досліджуваних для вивчення біоеквівалентності повинен бути якомога одноріднішим,
Вимоги до випробовуваних
Контингент досліджуваних для вивчення біоеквівалентності повинен бути якомога одноріднішим,

Слайд 16Можливість пацієнтів (здорових добровольців) брати участь в дослідженні повинна підтверджуватися даними, отриманими
Можливість пацієнтів (здорових добровольців) брати участь в дослідженні повинна підтверджуватися даними, отриманими

Слайд 17Кількість випробовуваних для проведення дослідження
Мінімальне число випробовуваних, таких, що залучаються до досліджень
Кількість випробовуваних для проведення дослідження
Мінімальне число випробовуваних, таких, що залучаються до досліджень

Слайд 18Особливості режиму для випробовуваних при проведенні дослідження
З метою мінімізації відмінностей, що виникають
Особливості режиму для випробовуваних при проведенні дослідження
З метою мінімізації відмінностей, що виникають

Слайд 19Особливості дизайну випробування
Особливістю дизайну таких досліджень є те, що кожний з
Особливості дизайну випробування
Особливістю дизайну таких досліджень є те, що кожний з

Слайд 20Фармкокінетичні параметри, які визначаються
для оцінки біодоступності і біоеквівалентності
При вивченні біодоступності
Фармкокінетичні параметри, які визначаються
для оцінки біодоступності і біоеквівалентності
При вивченні біодоступності

Слайд 21АС, мкг/мл
Основні параметри фармакокінетики, використовувані при вивченні біоеквівалентності лікарських речовин
АС, мкг/мл
Основні параметри фармакокінетики, використовувані при вивченні біоеквівалентності лікарських речовин
Слайд 22При вивченні біодоступності методом введення одноразової дози площа під кривою «концентрація-час» визначається
При вивченні біодоступності методом введення одноразової дози площа під кривою «концентрація-час» визначається

Слайд 23Час відбору проб для дослідження
Час узяття проб крові у випробовуваних необхідно
Час відбору проб для дослідження
Час узяття проб крові у випробовуваних необхідно

Слайд 24Значення показника максимальної концентрації речовини можна пояснити за допомогою наступного прикладу.
На
Значення показника максимальної концентрації речовини можна пояснити за допомогою наступного прикладу.
На

Слайд 25Зіставлення максимальних концентрацій в крові двох препаратів: МЕК - мінімальна ефективна концентрація;
Зіставлення максимальних концентрацій в крові двох препаратів: МЕК - мінімальна ефективна концентрація;
Слайд 26Крива 1 характеризує концентрацію в крові стандартного препарату, а крива 2 -
Крива 1 характеризує концентрацію в крові стандартного препарату, а крива 2 -

Слайд 27На малюнку 3 показано, що максимальна концентрація стандартного препарату (крива 1) досягається
На малюнку 3 показано, що максимальна концентрація стандартного препарату (крива 1) досягається

Слайд 28Зіставлення часу досягнення максимальної концентрації двох препаратів: МЕК - мінімальна ефективна концентрація;
Зіставлення часу досягнення максимальної концентрації двох препаратів: МЕК - мінімальна ефективна концентрація;
Слайд 29На малюнку 4 представлений інший приклад співвідношення кривих, що відображають кінетику двох
На малюнку 4 представлений інший приклад співвідношення кривих, що відображають кінетику двох

Слайд 30Зіставлення площ під фармакокинетичними кривими двох препаратів: 1 - стандартний препарат; 2
Зіставлення площ під фармакокинетичними кривими двох препаратів: 1 - стандартний препарат; 2

Слайд 31Таким чином, два препарати вважаються біоеквівалентними, якщо вони мають схожі фармакокінетичні показники.
Таким чином, два препарати вважаються біоеквівалентними, якщо вони мають схожі фармакокінетичні показники.

Слайд 32Звіт про проведене випробування
Всі етапи проведення дослідження повинні ретельно фіксуватися, аналізуватися, описуватися,
Звіт про проведене випробування
Всі етапи проведення дослідження повинні ретельно фіксуватися, аналізуватися, описуватися,

 Лечение повреждений легкого
Лечение повреждений легкого ЭКГ. Прогрессирование острого инфаркта миокарда
ЭКГ. Прогрессирование острого инфаркта миокарда Злокачественная мезотелиома плевры
Злокачественная мезотелиома плевры Топография таза и промежности. Операции на органах малого таза
Топография таза и промежности. Операции на органах малого таза Научный эксперимент: наследственные болезни человека
Научный эксперимент: наследственные болезни человека Музей дефектологии Тюменской области
Музей дефектологии Тюменской области Возможности применения адаптированной модели ухода В. Хендерсон
Возможности применения адаптированной модели ухода В. Хендерсон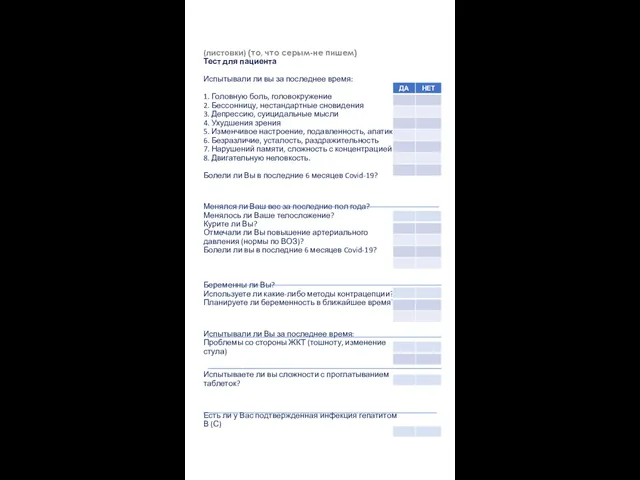 Тест для пациента
Тест для пациента Антигистаминные средства
Антигистаминные средства NPE
NPE Бронхиальная астма
Бронхиальная астма Комплексная помощь детям с дизартрией
Комплексная помощь детям с дизартрией Обеспечение медицинских организаций системы здравоохранения квалифицированными кадрами
Обеспечение медицинских организаций системы здравоохранения квалифицированными кадрами Постановка согревающего компресса. Постановка пузыря со льдом
Постановка согревающего компресса. Постановка пузыря со льдом Балалардың асқазанының сәулелі диагностикасы
Балалардың асқазанының сәулелі диагностикасы prezentatsia_povrezhdenia (1)
prezentatsia_povrezhdenia (1) Медицинский колледж. Акушерское дело
Медицинский колледж. Акушерское дело Энтеровирусный менингит
Энтеровирусный менингит Болезнь Паркинсона
Болезнь Паркинсона Клінічний випадок
Клінічний випадок Предотвратите пневмонию
Предотвратите пневмонию Тактика участкового терапевта при неотложных состояниях в эндокринологии. Лекция 26
Тактика участкового терапевта при неотложных состояниях в эндокринологии. Лекция 26 Инфекционные заболевания и их профилактика
Инфекционные заболевания и их профилактика ЛЕЧЕНИЕ, ПОДДЕРЖИВАНИЕ И ПРЕДОТВРАЩЕНИЕ в Пародонтологии. - презентация_
ЛЕЧЕНИЕ, ПОДДЕРЖИВАНИЕ И ПРЕДОТВРАЩЕНИЕ в Пародонтологии. - презентация_ Эктодермальные дисплазии. Определение. Классификация. Энтеропатический акродерматит. Определение
Эктодермальные дисплазии. Определение. Классификация. Энтеропатический акродерматит. Определение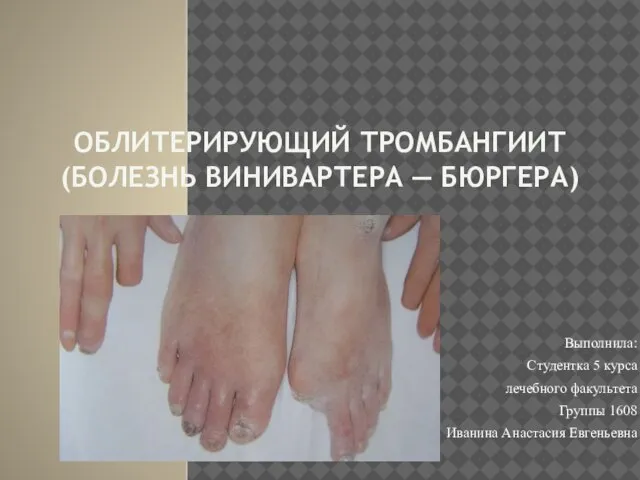 Облитерирующий тромбангиит (болезнь Винивартера — Бюргера)
Облитерирующий тромбангиит (болезнь Винивартера — Бюргера) Акушерский инструментарий. Операции из истории
Акушерский инструментарий. Операции из истории Диагностика и лечение новообразований
Диагностика и лечение новообразований