- Гломерулонефриты у детей и подростков

Содержание
- 2. Рост числа больных с хроническими болезнями почек Результаты лечения ряда хронических заболеваний почек не удовлетворяют Растет
- 3. Гломерулонефриты, или иммунные гломерулопатии, – это гетерогенная группа заболеваний, для которых характерно наличие иммунологических и клинико-морфологических
- 4. Для выделения нозологической формы ГН необходимо определить: 1.Синдром и динамику клинических симптомов 2.Характеристику иммунопатологических изменений 3.Морфологическую
- 5. Первичные По характеру течения Острый ГН Хронический ГН Быстропрогрессирующий ГН Вторичные при ряде системных заболеваний Гломерулонефриты
- 7. Острое диффузное иммуновоспалительное поражение почек, возникающее после бактериального, вирусного или паразитарного заболевания, спустя латентный период (2-3
- 8. Стрептококки гр.А, штамм 12 (60-80%), 1,3,4,49 Nephritis-associated plasmin receptor (NAPIr) – нефритогенный антиген стрептококка группы А.
- 9. Инфекции и заболевания, предшествовавшие ОГН: 1.Воспаление в носоглотке, на коже; 2.Бактериальный эндокардит 3.Пневмония 4.Менингит 5.Гепатит В
- 10. Предрасполагающие факторы: Антигенный набор HLA DR4, DR5 Отягощенная наследственность по инфекционно-аллергическим заболеваниям Высокая восприимчивость к стрептококковым
- 11. АГстрептококка + АТ + С3а,С5а → осаждение на базальной мембране клубочков Мембраноатакующий комплекс (С5в-С9) → ↑
- 12. Эндокапиллярный диффузный пролиферативный гломерулит; проходит несколько стадий: экссудативная, экссудативно-пролиферативная пролиферативная остаточных явлений Электронная микроскопия: «горбы» на
- 14. Отеки- ↓ СКФ, + ↑ реабсорбции Na = ↑ОЦК, задержка жидкости и Na в тканях +
- 15. Циклическое течение и нефритический синдром Через 2-3 недели после инфекции – отеки; м.б. олигурия (на 3-5
- 16. Предшествующая стрептококковая инфекция Латентный период 2-3 недели Острое начало, нефритический синдром Кратковременность нарушения функции почек В
- 17. ↑↑ АСЛ-О, АСК ↓ ↓ фракций С3 и С5; ↑ ЦИК В крови лейкоцитоз, нейтрофилез, ↑СОЭ,
- 18. Основная цель – уменьшить ишемию почек. Режим постельный на 3-4 недели Диета: соль, жидкость, белок, калорийность
- 19. В 85-90% - выздоровление. Факторы прогрессирования – интерстициальные изменения ↓ уд. плотности мочи, лейкоцитурия, ↓ осмотического
- 20. ХГН – первичное вовлечение в иммунопатологическое воспаление клубочков с последующим поражением канальцев, интерстиция, с развитием в
- 21. У детей первых 5-6 лет преобладает ГН с минимальными изменениями 60-70% ХГН – мезангиопролиферативный (течение относительно
- 22. Мезангипролиферативный гломерулит- наиболее частая форма у младших школьников
- 24. Быстропрогрессирующий ГН с полулуниями – типично для подростков
- 25. Мембранозный гломерулонефрит- редкая форма у детей
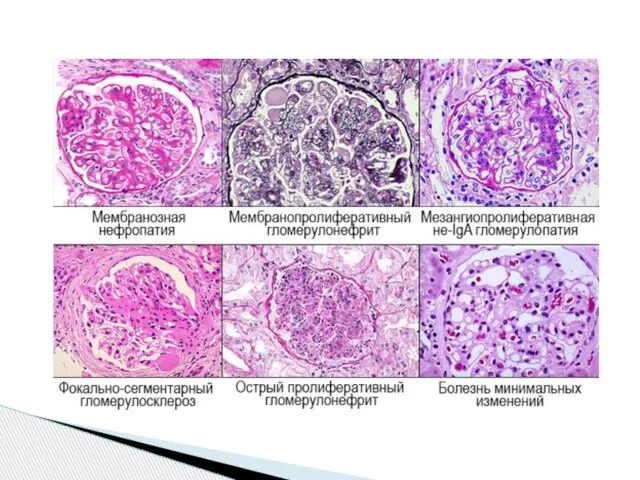
- 26. Форма (синдром): 1.нефритическая (гематурическая) 2.нефротическая 3.смешанная Период 1.Обострение 2.Частичная ремиссия 3.Полная клинико-лабораторная ремиссия Функция почек 1.Без
- 27. Форма нефритическая или гематурическая – морфологически мезангиально -пролиферативный или пролиферативно-мембранозный ГН. В клинике – умеренная гематурия
- 28. Форма нефротическая – морфологически минимальный ГН, мембранозный, мембранозно -пролиферативный ГН. В клинике – нефротический синдром. АД
- 29. Форма смешанная –пролиферативно-мембранозный и пролиферативно-фибропластический ГН. В клинике – упорные длительные отёки, АГ с изменениями глазного
- 30. Характеризуется Относительно быстрым началом (в течение 1-2 недель) Протеинурией (возможно нефротического уровня), гематурией (микро-,макро-), цилиндрурией Прогрессирующим
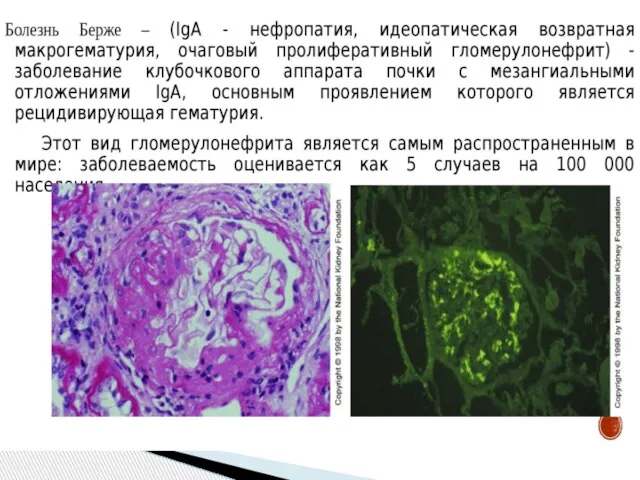
- 31. Болезнь Берже, IgA-нефропатия Код по МКБ-10 N02 – рецидивирующая и устойчивая гематурия
- 32. Распространенность: наиболее частая ф. Первичного ГН 44,4% детей с гематурической ф.ГН 12,1% детей и 12,6% взрослых,
- 34. Причины Этиология • Вирусы гепатита В, герпесвирусы • Бактерии — E. coli, грибы, палочка Коха •
- 35. IgA вырабатывается в основном слизистыми; только 1/3 – лимфоцитами. Мономер IgA – из 2 тяжелых цепей
- 36. Выделяют 2 изотипа IgA – IgA1 и IgA2 IgA2 резистентен к бактериальным протеазам; кроме того плазматические
- 37. 3 этапа развития почечного повреждения: Депозиция IgA в мезангиуме Развитие повреждения мезангиума из-за взаимодействия IgA1-содержащих комплексов
- 38. 1.Рецидивирующая макрогематурия 2.Единственный эпизод макрогематурией с последующей персистенцией микрогематурии 3.Бессимптомная микрогематурия + протеинурия ( 4.Возможно развитие
- 39. Склеротические изменения клубочков ---»---»---» в сочетании пролиферации мезангия со склерозом 20% гломерул Гломерулярные полулуния Артериальная гипертензия
- 40. Клинически – дифференциальный диагноз с МКБ, геморрагическими циститами, онкопатологией мочевого пузыря Иммунофлюоресцентное изучение биоптата При отсутствии
- 41. • Иногда эффективны антибиотикотерапия или изменение диеты • Больным с изолированной гематурией назначают ингибиторы АПФ или
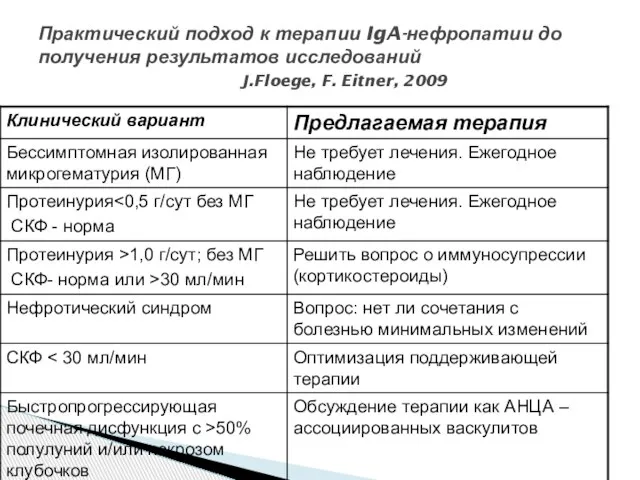
- 42. Практический подход к терапии IgA-нефропатии до получения результатов исследований J.Floege, F. Eitner, 2009
- 43. Течение рецидивирующее Исход в ХПН – через 10-15 лет Возможна спонтанная ремиссия. У детей прогноз лучше,
- 44. Вторичный гломерулонефрит
- 45. Люпус-нефрит и гематологический синдром при СКВ – ведущие в определении тяжести, инвалидизации и прогноза. Среди основных
- 46. Симптомокомплекс, при котором поражаются многие системы и органы, характеризуется наличием в циркуляции антител, направленных против одного
- 47. В запуске патологического процесса при СКВ приписывается роль инфекционным агентам (туберкулезная палочка, ретровирус, E. сoli…) Существует
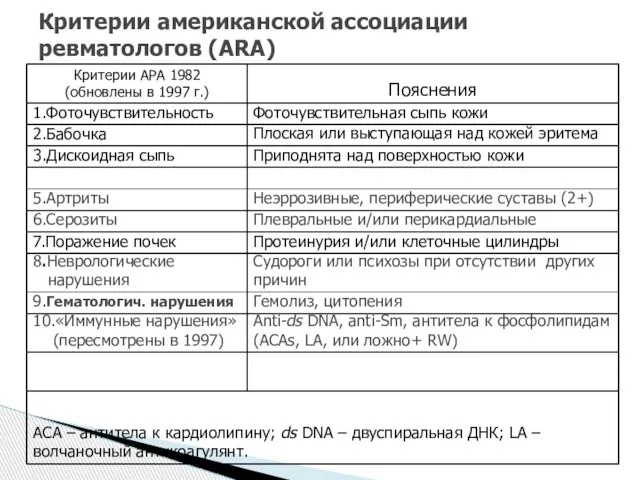
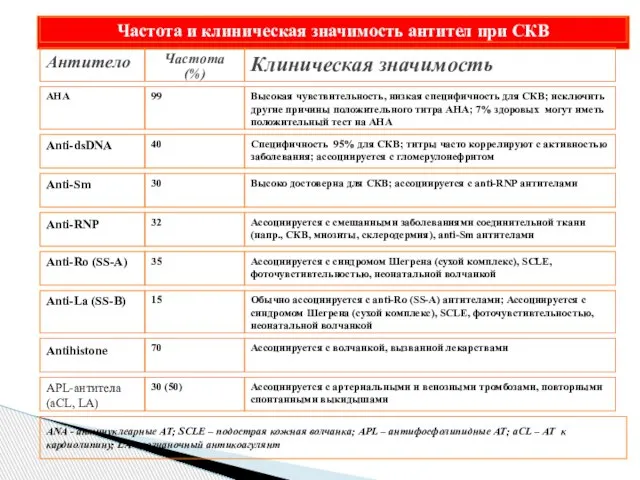
- 48. Критерии американской ассоциации ревматологов (ARA)
- 50. Волчаночный нефрит ISN & RPS Working group on the classification of LN, JASN 2003 15:241-250
- 51. Поражаются сосуды; утолщение БМ клубочка; Гломерулосклероз; ↓ почечного кровотока. Ангиографически: констрикция внутридольковых артерий (тот же процесс,
- 52. Генетически детерминированные неиммунные гломерулопатии, протекающие с гематурией, прогрессирующим снижением почечных функций Частота НН (По России) 17
- 53. Генетическая основа – мутация в гене α-5-цепи коллагена IV типа Коллаген IV типа универсален для базальных
- 54. 3 клинических варианта: Синдром Альпорта НН без тугоухости Семейная доброкачественная гематурия Наследственный нефрит
- 55. СИНДРОМ АЛЬПОРТА Синдром Альпорта наследственное (обычно сцепленное с Х-хромосомой) заболевание, характеризующееся патологией гломерул и часто ассоциирующееся
- 56. СИНДРОМ АЛЬПОРТА Первое описание семьи, в которой наблюдались случаи гематурии в нескольких поколениях принадлежит L. Guthrie
- 57. СИНДРОМ АЛЬПОРТА Синдром Альпорта причина терминальной почечной недостаточности у 2,5% детей и 0,3% взрослых Частота синдрома
- 58. СИНДРОМ АЛЬПОРТА В основе генетический дефект приводящий к патологии коллагена IV типа, входящего в состав базальных
- 59. СИНДРОМ АЛЬПОРТА (морфология) При световой микроскопии изменения не специфичны У маленьких детей ( В более старшем
- 60. СИНДРОМ АЛЬПОРТА (морфология) Иммунофлюоресцентное исследование, как правило, негативно Изредка выявляются отложения С3 и IgM – различной
- 61. СИНДРОМ АЛЬПОРТА (морфология) Электронная микроскопия В начальных стадиях заболевания может выявляться только утончение ГБМ, практически не
- 62. Ультраструктурные изменения ГБМ при синдроме Альпорта. БМ утолщена, слоиста, с неровными контурами (ув. Х 9000)
- 63. Первые симптомы – в первые 3 года жизни, случайно при исследовании анализа мочи Гематурия стойкая, разной
- 64. СИНДРОМ АЛЬПОРТА (клиника - 2) Возможно развитие нефротического синдрома Гипертензия, как правило, выявляется в поздних стадиях
- 65. СИНДРОМ АЛЬПОРТА (клиника-3) Частота выявления нейросенсорной глухоты составляет 30-50% Нарушения слуха всегда сопровождаются патологией почек Тяжесть
- 66. Диагностика наследственного нефрита (синдрома Альпорта) Необходимо наличие трех из следующих пяти признаков: гематурия или летальный исход
- 67. Диагностика наследственного нефрита (синдрома Альпорта) Генетический скрининг [синдрома Альпорта] затруднен из-за наличия большого числа мутаций и
- 68. Не разработано Ренопротекция (малобелковая диета, ингибиторы АПФ, коррекция артериальной гипертензии) Заместительная почечная терапия (гемодиализ, трансплантация почки*)
- 69. БТБМ рассматривается как состояние, характеризующееся утончением ГБМ при электронной микроскопии, клинически проявляющееся изолированной гематурией, часто наблюдающейся
- 70. Тем не менее при длительном наблюдении у 30-35% пациентов с БТБМ может выявляться артериальная гипертензия Болезнь
- 71. Генетические исследования свидетельствуют о том, что БТБМ - генетически гетерогенное заболевание, которое чаще наследуется по аутосомно-доминатному
- 72. БТБМ, по видимому, не является очень редким заболеванием, поскольку ее признаки при электронномикроскопическом исследовании биоптата могут
- 73. Частота выявления БТБМ, по видимому, увеличивается с возрастом и это заболевание чаще встречается у мужчин, чем
- 74. Абсолютно четкой грани между синдромом Альпорта и БТБМ тонких мембран в настоящее время провести нельзя! И.К.
- 75. Эпизодические головокружения, чаще в вечернее время, без четкой связи с физической нагрузкой. Эпизодические подъемы АД до
- 76. С возраста 1 г отмечается микрогематурия (1-6 в п/зр) С 14 лет нарастание гематурии до 40-50
- 77. Показатели клинического анализа крови
- 78. Показатели функционального состояние почек при поступлении
- 79. Световая микроскопия: В срезах мозговой и корковый слой с числом клубочков до 22. Клубочки средних размеров
- 80. Иммунофлюоресцентное исследование биоптата почки Заключение: В клубочках и тубулоинтерстициальной системе почки отложений иммуноглобулинов и компонентов комплемента
- 83. Болезнь тонкой базальной мембраны с мезангиальной пролиферацией. Сохранная функция почек. Диагноз:
- 84. Не менее 2 больных нефропатией в семье Гематурия как ведущий симптом нефропатии у пробанда Наличие тугоухости
- 85. Гематурическая форма гломерулонефрита Болезнь Берже Дисметаболическая нефропатия Дифференциальная диагностика наследственного нефрита
- 86. Генетическое обследование Полноценное питание АТФ, кокарбоксилаза, пиридоксин, В15, карнитина хлорид. (Курсы 2 – 3 раза в
- 87. Нефротический синдром Липоидный нефроз, или гломерулонефрит с минимальными изменениями
- 88. Нефротический синдром (НС) - симптомокомплекс, для которого характерны: протеинурия у детей не менее 50 мг/кг/24ч гипоальбуминемия
- 89. В основе НС лежит протеинурия, зависящая от нарушений белков, подоцитов и подоцитарной диафрагмы
- 90. Строение подоцита H. Pavenstädt, W. Kriz, and M. Kretzler, 2003; Weiming Yu et al., 2005
- 91. Строение подоцита H. Pavenstädt, W. Kriz, and M. Kretzler, 2003;
- 92. Белки щелевой мембраны Marilyn G. Farquhar, 2006; H. Pavenstädt, W. Kriz, and M. Kretzler, 2003
- 93. Белки подоцитов и щелевой мембраны Нефрин Подоцин ZO1-мембранный белок CD2AP (CD2-ассоциированный протеин) Синаптоподин Деснин FAT-трансмембранный адгезивный
- 94. Функции подоцитов Синтез белков гломерулярной базальной мембраны Регулирование растяжимости клубочкового капилляра Ограничение попадания в мочевое пространство
- 95. В основе НС лежит протеинурия, зависящая от нарушений белков, подоцитов и подоцитарной диафрагмы Симптомокомплекс: протеинурия +
- 96. Нефротический синдром Протеинурия – первична У взрослых – 3,5 г/сутки У детей: 1г/(м² х сут.) более
- 97. Нефротический синдром Гипоальбуминемия при НС 30 – 25 г/л Нарушения липидного обмена – 3 типа: Усиление
- 98. Нефротический синдром Отеки в зависимости от внутрисосудистого объема жидкости: гиперволемические, гиповолемические нормоволемические При НС с минимальными
- 99. Врожденный и инфантильный НС 1.Первичный: Врожденный НС финского типа с микрокистозом Врожденный НС, французский тип с
- 100. Врожденный НС финского типа Finnish Type N.S., finnisher Type NS, неонатальный нефроз Часто – в Финляндии
- 101. НС с минимальными изменениями – липоидный нефроз у детей НС с минимальными измен. Липоидный нефроз Идиопатический
- 102. НС с минимальными изменениями – липоидный нефроз у детей Возраст – 1,5 – 7 лет Мальчики
- 103. Нефротический синдром - определение НС, преобладающий в структуре НС у детей 1-14 лет,: Начало заболевания с
- 104. НС с минимальными изменениями – липоидный нефроз у детей Клиника: отеки при удовлетворительном состоянии (рыхлые мягкие,
- 105. НС с минимальными изменениями – липоидный нефроз у детей Диагноз в 90-95% - по клинике +
- 106. Нефротический синдром – варианты течения Гормоночувствительный НС без рецидивов: нормализация мочи в течение 4 – 8
- 107. НС с минимальными изменениями – липоидный нефроз у детей Течение и исходы: Острое → стойкая клинико-лабораторная
- 108. Нефротический синдром – стероидорезистентность Отсутствие эффекта от преднизолона в максимальной дозе 60 мг/м²/сут в течение 4-8
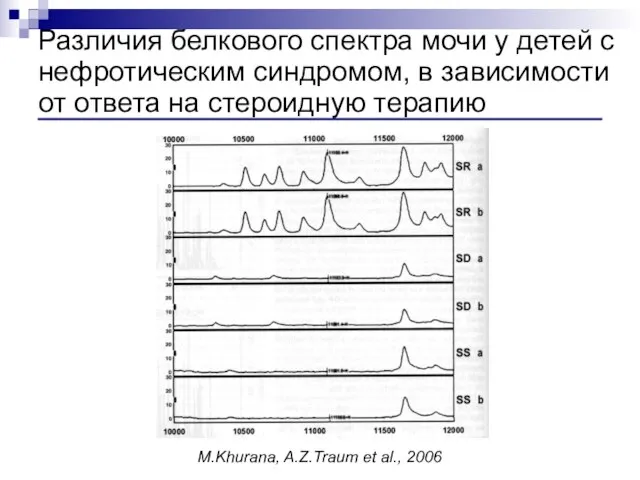
- 109. Различия белкового спектра мочи у детей с нефротическим синдромом, в зависимости от ответа на стероидную терапию
- 110. НС с минимальными изменениями – липоидный нефроз у детей - лечение Режим: не ограничивать движения (риск
- 111. Нефротический синдром – стероидная зависимость Рецидив НС при снижении дозы или полной отмене преднизолона Возникновение рецидивов
- 112. НС с минимальными изменениями – липоидный нефроз у детей Побочные эффекты преднизолона (стероидотоксичность) Синдром Иценко-Кушинга Гипокальциемия,
- 113. Нефротический синдром – показания к цитостатической терапии Гормонорезистентность в дебюте заболевания Часто рецидивирующее течение Развитие стероидной
- 114. НС с минимальными изменениями – липоидный нефроз у детей - осложнения Нефротический криз – анасарка, альбумин
- 115. НС с минимальными изменениями – липоидный нефроз у детей - осложнения Почечная эклампсия (ангиоспастическая энцефалопатия) –
- 116. Фокально-сегментарный гломерулосклероз (ФСГС) – диагноз – на данных морфологических исследований Форма гломерулопатии, для которой характерно склерозирование
- 117. ФСГС ФСГС – самая частая причина НС у взрослых (20-25%) И стероидрезистентного НС у детей (более
- 118. ФСГС у детей - лечение 1.После нефробиопсии при стероидрезистентности 2.Детям раннего возраста с субнефротической протеинурией и
- 119. НС с минимальными изменениями – цитостатическая терапия Алкилирующие препараты: (детям не рекомендуются) Хлорбутин 0,15-0,3мг/кг/сут или 0,1-0.2
- 121. Скачать презентацию
Слайд 2Рост числа больных с хроническими болезнями почек
Результаты лечения ряда хронических заболеваний почек
Рост числа больных с хроническими болезнями почек
Результаты лечения ряда хронических заболеваний почек

Слайд 3Гломерулонефриты,
или иммунные гломерулопатии, – это
гетерогенная группа заболеваний,
для которых характерно
наличие иммунологических
Гломерулонефриты,
или иммунные гломерулопатии, – это
гетерогенная группа заболеваний,
для которых характерно
наличие иммунологических

Слайд 4Для выделения нозологической формы ГН
необходимо определить:
1.Синдром и динамику клинических симптомов
2.Характеристику иммунопатологических изменений
3.Морфологическую
Для выделения нозологической формы ГН
необходимо определить:
1.Синдром и динамику клинических симптомов
2.Характеристику иммунопатологических изменений
3.Морфологическую

Слайд 5Первичные
По характеру течения
Острый ГН
Хронический ГН
Быстропрогрессирующий ГН
Вторичные
при ряде системных
заболеваний
Гломерулонефриты у детей
Первичные
По характеру течения
Острый ГН
Хронический ГН
Быстропрогрессирующий ГН
Вторичные
при ряде системных
заболеваний
Гломерулонефриты у детей


Слайд 7Острое диффузное иммуновоспалительное поражение почек, возникающее после бактериального, вирусного или паразитарного заболевания,
Острое диффузное иммуновоспалительное поражение почек, возникающее после бактериального, вирусного или паразитарного заболевания,

Слайд 8Стрептококки гр.А, штамм 12 (60-80%), 1,3,4,49
Nephritis-associated plasmin receptor (NAPIr) – нефритогенный антиген
Стрептококки гр.А, штамм 12 (60-80%), 1,3,4,49
Nephritis-associated plasmin receptor (NAPIr) – нефритогенный антиген

Слайд 9Инфекции и заболевания, предшествовавшие ОГН:
1.Воспаление в носоглотке, на коже;
2.Бактериальный эндокардит
3.Пневмония
4.Менингит
5.Гепатит В
6.Вирус Эпштейн-Барра
7.Цитомегаловирус
8.Вирус
Инфекции и заболевания, предшествовавшие ОГН:
1.Воспаление в носоглотке, на коже;
2.Бактериальный эндокардит
3.Пневмония
4.Менингит
5.Гепатит В
6.Вирус Эпштейн-Барра
7.Цитомегаловирус
8.Вирус

Слайд 10Предрасполагающие факторы:
Антигенный набор HLA DR4, DR5
Отягощенная наследственность по инфекционно-аллергическим заболеваниям
Высокая восприимчивость к
Предрасполагающие факторы:
Антигенный набор HLA DR4, DR5
Отягощенная наследственность по инфекционно-аллергическим заболеваниям
Высокая восприимчивость к

Слайд 11АГстрептококка + АТ + С3а,С5а → осаждение на базальной мембране клубочков
Мембраноатакующий комплекс
АГстрептококка + АТ + С3а,С5а → осаждение на базальной мембране клубочков
Мембраноатакующий комплекс

Слайд 12Эндокапиллярный диффузный пролиферативный
гломерулит; проходит несколько стадий:
экссудативная,
экссудативно-пролиферативная
пролиферативная
остаточных явлений
Электронная
Эндокапиллярный диффузный пролиферативный
гломерулит; проходит несколько стадий:
экссудативная,
экссудативно-пролиферативная
пролиферативная
остаточных явлений
Электронная


Слайд 14Отеки- ↓ СКФ, + ↑ реабсорбции Na = ↑ОЦК, задержка жидкости и
Отеки- ↓ СКФ, + ↑ реабсорбции Na = ↑ОЦК, задержка жидкости и

Слайд 15Циклическое течение и нефритический синдром
Через 2-3 недели после инфекции – отеки; м.б.
Циклическое течение и нефритический синдром
Через 2-3 недели после инфекции – отеки; м.б.

Слайд 16Предшествующая стрептококковая инфекция
Латентный период 2-3 недели
Острое начало, нефритический синдром
Кратковременность нарушения функции
Предшествующая стрептококковая инфекция
Латентный период 2-3 недели
Острое начало, нефритический синдром
Кратковременность нарушения функции

Слайд 17↑↑ АСЛ-О, АСК
↓ ↓ фракций С3 и С5; ↑ ЦИК
В крови лейкоцитоз,
↑↑ АСЛ-О, АСК
↓ ↓ фракций С3 и С5; ↑ ЦИК
В крови лейкоцитоз,

Слайд 18Основная цель – уменьшить ишемию почек.
Режим постельный на 3-4 недели
Диета: соль, жидкость,
Основная цель – уменьшить ишемию почек.
Режим постельный на 3-4 недели
Диета: соль, жидкость,

Слайд 19В 85-90% - выздоровление.
Факторы прогрессирования – интерстициальные изменения ↓ уд. плотности мочи,
В 85-90% - выздоровление.
Факторы прогрессирования – интерстициальные изменения ↓ уд. плотности мочи,

Слайд 20ХГН – первичное вовлечение в иммунопатологическое воспаление клубочков с последующим поражением канальцев,
ХГН – первичное вовлечение в иммунопатологическое воспаление клубочков с последующим поражением канальцев,

Слайд 21У детей первых 5-6 лет преобладает ГН с минимальными изменениями
60-70% ХГН –
У детей первых 5-6 лет преобладает ГН с минимальными изменениями
60-70% ХГН –

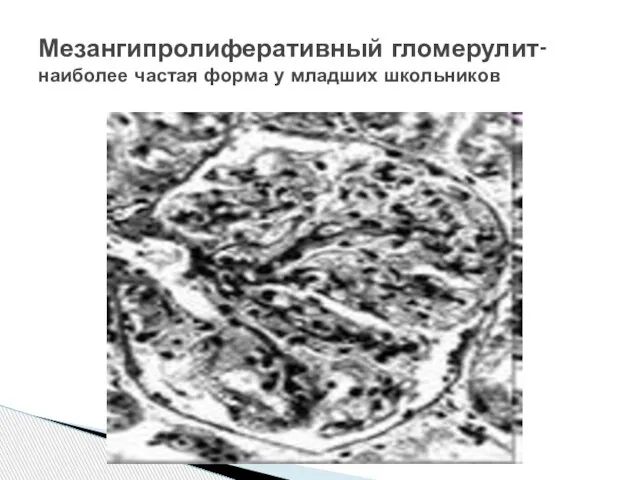
Слайд 22Мезангипролиферативный гломерулит-
наиболее частая форма у младших школьников
Мезангипролиферативный гломерулит-
наиболее частая форма у младших школьников
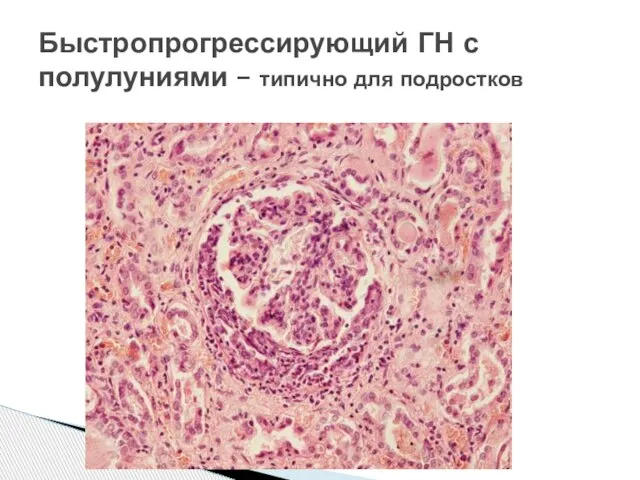
Слайд 24Быстропрогрессирующий ГН с полулуниями – типично для подростков
Быстропрогрессирующий ГН с полулуниями – типично для подростков
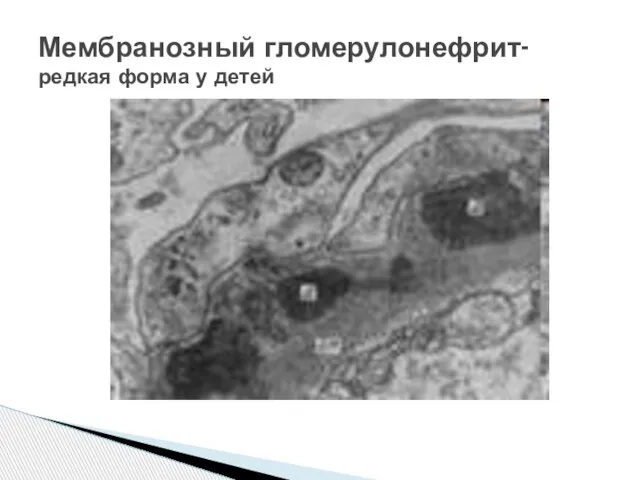
Слайд 25Мембранозный гломерулонефрит-
редкая форма у детей
Мембранозный гломерулонефрит-
редкая форма у детей
Слайд 26Форма (синдром):
1.нефритическая (гематурическая)
2.нефротическая
3.смешанная
Период
1.Обострение
2.Частичная ремиссия
3.Полная клинико-лабораторная ремиссия
Функция
Форма (синдром):
1.нефритическая (гематурическая)
2.нефротическая
3.смешанная
Период
1.Обострение
2.Частичная ремиссия
3.Полная клинико-лабораторная ремиссия
Функция

Слайд 27Форма нефритическая или гематурическая – морфологически мезангиально -пролиферативный или пролиферативно-мембранозный ГН.
В
Форма нефритическая или гематурическая – морфологически мезангиально -пролиферативный или пролиферативно-мембранозный ГН.
В

Слайд 28Форма нефротическая – морфологически минимальный ГН, мембранозный, мембранозно -пролиферативный ГН. В клинике
Форма нефротическая – морфологически минимальный ГН, мембранозный, мембранозно -пролиферативный ГН. В клинике

Слайд 29Форма смешанная –пролиферативно-мембранозный и пролиферативно-фибропластический ГН. В клинике – упорные длительные отёки,
Форма смешанная –пролиферативно-мембранозный и пролиферативно-фибропластический ГН. В клинике – упорные длительные отёки,

Слайд 30Характеризуется
Относительно быстрым началом (в течение 1-2 недель)
Протеинурией (возможно нефротического уровня), гематурией (микро-,макро-),
Характеризуется
Относительно быстрым началом (в течение 1-2 недель)
Протеинурией (возможно нефротического уровня), гематурией (микро-,макро-),

Слайд 31Болезнь Берже,
IgA-нефропатия
Код по МКБ-10 N02 – рецидивирующая и устойчивая гематурия
Болезнь Берже,
IgA-нефропатия
Код по МКБ-10 N02 – рецидивирующая и устойчивая гематурия

Слайд 32Распространенность: наиболее частая ф. Первичного ГН
44,4% детей с гематурической ф.ГН
12,1% детей и
Распространенность: наиболее частая ф. Первичного ГН
44,4% детей с гематурической ф.ГН
12,1% детей и

Слайд 34Причины
Этиология • Вирусы гепатита В, герпесвирусы • Бактерии — E. coli, грибы,
Причины
Этиология • Вирусы гепатита В, герпесвирусы • Бактерии — E. coli, грибы,

Слайд 35IgA вырабатывается в основном слизистыми; только 1/3 – лимфоцитами.
Мономер IgA – из
IgA вырабатывается в основном слизистыми; только 1/3 – лимфоцитами.
Мономер IgA – из

Слайд 36Выделяют 2 изотипа IgA – IgA1 и IgA2
IgA2 резистентен к бактериальным протеазам;
Выделяют 2 изотипа IgA – IgA1 и IgA2
IgA2 резистентен к бактериальным протеазам;

Слайд 373 этапа развития почечного повреждения:
Депозиция IgA в мезангиуме
Развитие повреждения мезангиума из-за взаимодействия
3 этапа развития почечного повреждения:
Депозиция IgA в мезангиуме
Развитие повреждения мезангиума из-за взаимодействия

Слайд 381.Рецидивирующая макрогематурия
2.Единственный эпизод макрогематурией с последующей персистенцией микрогематурии
3.Бессимптомная микрогематурия + протеинурия
1.Рецидивирующая макрогематурия
2.Единственный эпизод макрогематурией с последующей персистенцией микрогематурии
3.Бессимптомная микрогематурия + протеинурия

Слайд 39Склеротические изменения клубочков
---»---»---» в сочетании пролиферации мезангия со склерозом 20% гломерул
Гломерулярные полулуния
Склеротические изменения клубочков
---»---»---» в сочетании пролиферации мезангия со склерозом 20% гломерул
Гломерулярные полулуния

Слайд 40Клинически – дифференциальный диагноз с МКБ, геморрагическими циститами, онкопатологией мочевого пузыря
Иммунофлюоресцентное изучение
Клинически – дифференциальный диагноз с МКБ, геморрагическими циститами, онкопатологией мочевого пузыря
Иммунофлюоресцентное изучение

Слайд 41• Иногда эффективны антибиотикотерапия или изменение диеты
• Больным с изолированной гематурией
• Иногда эффективны антибиотикотерапия или изменение диеты
• Больным с изолированной гематурией

Слайд 42Практический подход к терапии IgA-нефропатии до получения результатов исследований
J.Floege, F. Eitner,
Практический подход к терапии IgA-нефропатии до получения результатов исследований J.Floege, F. Eitner,
Слайд 43Течение рецидивирующее
Исход в ХПН – через 10-15 лет
Возможна спонтанная ремиссия.
У детей прогноз
Течение рецидивирующее
Исход в ХПН – через 10-15 лет
Возможна спонтанная ремиссия.
У детей прогноз

Слайд 44Вторичный гломерулонефрит
Вторичный гломерулонефрит

Слайд 45Люпус-нефрит и гематологический синдром при СКВ – ведущие в определении тяжести, инвалидизации
Люпус-нефрит и гематологический синдром при СКВ – ведущие в определении тяжести, инвалидизации

Слайд 46Симптомокомплекс, при котором поражаются многие системы и органы, характеризуется наличием в циркуляции
Симптомокомплекс, при котором поражаются многие системы и органы, характеризуется наличием в циркуляции

Слайд 47В запуске патологического процесса при СКВ приписывается роль инфекционным агентам (туберкулезная палочка,
В запуске патологического процесса при СКВ приписывается роль инфекционным агентам (туберкулезная палочка,

Слайд 48Критерии американской ассоциации ревматологов (ARA)
Критерии американской ассоциации ревматологов (ARA)
Слайд 50Волчаночный нефрит
ISN & RPS Working group on the classification of LN,
Волчаночный нефрит
ISN & RPS Working group on the classification of LN,

Слайд 51Поражаются сосуды; утолщение БМ клубочка;
Гломерулосклероз; ↓ почечного кровотока.
Ангиографически: констрикция внутридольковых артерий (тот
Поражаются сосуды; утолщение БМ клубочка;
Гломерулосклероз; ↓ почечного кровотока.
Ангиографически: констрикция внутридольковых артерий (тот

Слайд 52Генетически детерминированные неиммунные гломерулопатии, протекающие с гематурией, прогрессирующим снижением почечных функций
Частота
Генетически детерминированные неиммунные гломерулопатии, протекающие с гематурией, прогрессирующим снижением почечных функций
Частота

Слайд 53Генетическая основа – мутация в гене
α-5-цепи коллагена IV типа
Коллаген
Генетическая основа – мутация в гене
α-5-цепи коллагена IV типа
Коллаген

Слайд 543 клинических варианта:
Синдром Альпорта
НН без тугоухости
Семейная доброкачественная
гематурия
Наследственный нефрит
3 клинических варианта:
Синдром Альпорта
НН без тугоухости
Семейная доброкачественная
гематурия
Наследственный нефрит

Слайд 55СИНДРОМ АЛЬПОРТА
Синдром Альпорта наследственное (обычно сцепленное с Х-хромосомой) заболевание, характеризующееся патологией гломерул
СИНДРОМ АЛЬПОРТА
Синдром Альпорта наследственное (обычно сцепленное с Х-хромосомой) заболевание, характеризующееся патологией гломерул

Слайд 56СИНДРОМ АЛЬПОРТА
Первое описание семьи, в которой наблюдались случаи гематурии в нескольких поколениях
СИНДРОМ АЛЬПОРТА
Первое описание семьи, в которой наблюдались случаи гематурии в нескольких поколениях

Слайд 57СИНДРОМ АЛЬПОРТА
Синдром Альпорта причина терминальной почечной недостаточности у 2,5% детей и 0,3%
СИНДРОМ АЛЬПОРТА
Синдром Альпорта причина терминальной почечной недостаточности у 2,5% детей и 0,3%

Слайд 58СИНДРОМ АЛЬПОРТА
В основе генетический дефект приводящий к патологии коллагена IV типа, входящего
СИНДРОМ АЛЬПОРТА
В основе генетический дефект приводящий к патологии коллагена IV типа, входящего

Слайд 59СИНДРОМ АЛЬПОРТА
(морфология)
При световой микроскопии изменения не специфичны
У маленьких детей (< 5 лет)
СИНДРОМ АЛЬПОРТА
(морфология)
При световой микроскопии изменения не специфичны
У маленьких детей (< 5 лет)

Слайд 60СИНДРОМ АЛЬПОРТА
(морфология)
Иммунофлюоресцентное исследование, как правило, негативно
Изредка выявляются отложения С3 и IgM –
СИНДРОМ АЛЬПОРТА
(морфология)
Иммунофлюоресцентное исследование, как правило, негативно
Изредка выявляются отложения С3 и IgM –

Слайд 61СИНДРОМ АЛЬПОРТА
(морфология)
Электронная микроскопия
В начальных стадиях заболевания может выявляться только утончение ГБМ, практически
СИНДРОМ АЛЬПОРТА
(морфология)
Электронная микроскопия
В начальных стадиях заболевания может выявляться только утончение ГБМ, практически

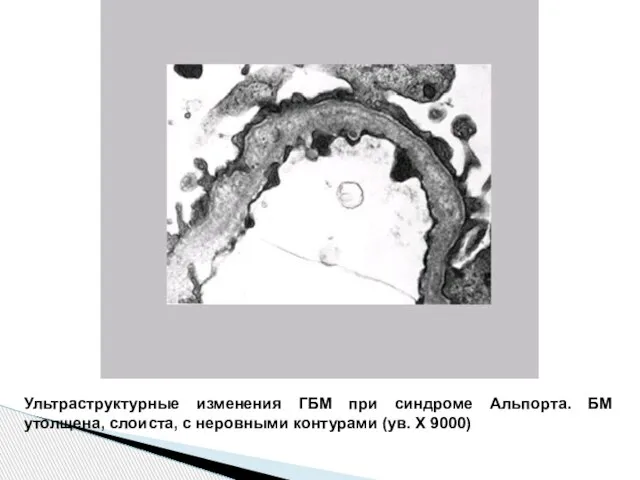
Слайд 62Ультраструктурные изменения ГБМ при синдроме Альпорта. БМ утолщена, слоиста, с неровными контурами
Ультраструктурные изменения ГБМ при синдроме Альпорта. БМ утолщена, слоиста, с неровными контурами
Слайд 63Первые симптомы – в первые 3 года жизни, случайно при исследовании анализа
Первые симптомы – в первые 3 года жизни, случайно при исследовании анализа

Слайд 64СИНДРОМ АЛЬПОРТА
(клиника - 2)
Возможно развитие нефротического синдрома
Гипертензия, как правило, выявляется в поздних
СИНДРОМ АЛЬПОРТА
(клиника - 2)
Возможно развитие нефротического синдрома
Гипертензия, как правило, выявляется в поздних

Слайд 65СИНДРОМ АЛЬПОРТА
(клиника-3)
Частота выявления нейросенсорной глухоты составляет 30-50%
Нарушения слуха всегда сопровождаются патологией почек
Тяжесть
СИНДРОМ АЛЬПОРТА
(клиника-3)
Частота выявления нейросенсорной глухоты составляет 30-50%
Нарушения слуха всегда сопровождаются патологией почек
Тяжесть

Слайд 66Диагностика наследственного нефрита (синдрома Альпорта)
Необходимо наличие трех из следующих пяти признаков:
гематурия
Диагностика наследственного нефрита (синдрома Альпорта)
Необходимо наличие трех из следующих пяти признаков:
гематурия

Слайд 67Диагностика наследственного нефрита (синдрома Альпорта)
Генетический скрининг [синдрома Альпорта] затруднен из-за наличия большого
Диагностика наследственного нефрита (синдрома Альпорта)
Генетический скрининг [синдрома Альпорта] затруднен из-за наличия большого
![Диагностика наследственного нефрита (синдрома Альпорта) Генетический скрининг [синдрома Альпорта] затруднен из-за наличия](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/896219/slide-66.jpg)
Слайд 68Не разработано
Ренопротекция (малобелковая диета, ингибиторы АПФ, коррекция артериальной гипертензии)
Заместительная почечная терапия (гемодиализ,
Не разработано
Ренопротекция (малобелковая диета, ингибиторы АПФ, коррекция артериальной гипертензии)
Заместительная почечная терапия (гемодиализ,

Слайд 69БТБМ рассматривается как состояние, характеризующееся утончением ГБМ при электронной микроскопии, клинически проявляющееся

Слайд 70
Тем не менее при длительном наблюдении у 30-35% пациентов с БТБМ может
Тем не менее при длительном наблюдении у 30-35% пациентов с БТБМ может

Слайд 71Генетические исследования свидетельствуют о том, что БТБМ - генетически гетерогенное заболевание, которое

Слайд 72БТБМ, по видимому, не является очень редким заболеванием, поскольку ее признаки при

Слайд 73Частота выявления БТБМ, по видимому, увеличивается с возрастом и это заболевание чаще

Слайд 74Абсолютно четкой грани между синдромом Альпорта и БТБМ тонких мембран в настоящее

Слайд 75 Эпизодические головокружения, чаще в вечернее время, без четкой связи с физической
Эпизодические головокружения, чаще в вечернее время, без четкой связи с физической

Слайд 76С возраста 1 г отмечается микрогематурия (1-6 в п/зр)
С 14 лет нарастание
С возраста 1 г отмечается микрогематурия (1-6 в п/зр)
С 14 лет нарастание

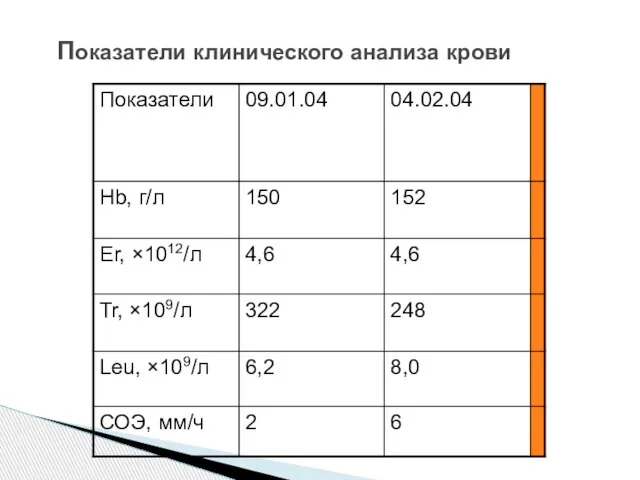
Слайд 77 Показатели клинического анализа крови
Показатели клинического анализа крови
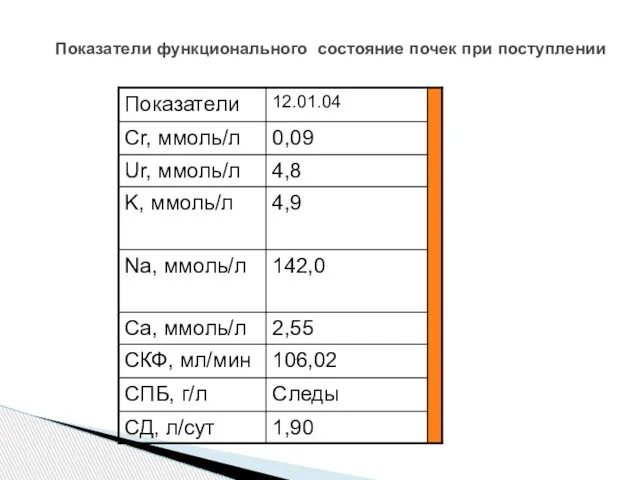
Слайд 78Показатели функционального состояние почек при поступлении
Показатели функционального состояние почек при поступлении
Слайд 79Световая микроскопия:
В срезах мозговой и корковый слой с числом клубочков до
Световая микроскопия:
В срезах мозговой и корковый слой с числом клубочков до

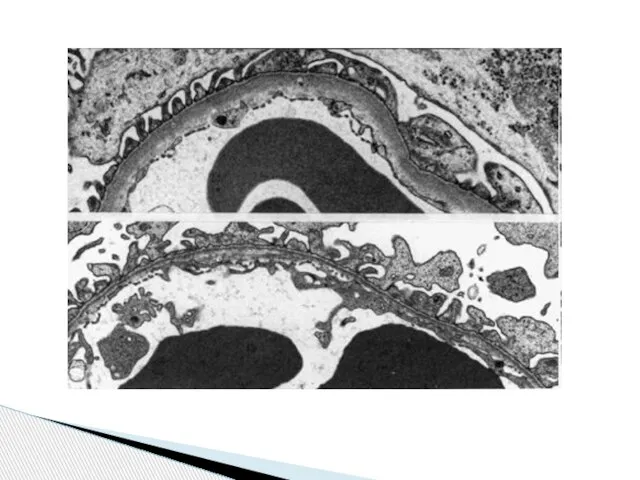
Слайд 80Иммунофлюоресцентное исследование биоптата почки
Заключение: В клубочках и тубулоинтерстициальной системе почки отложений
Иммунофлюоресцентное исследование биоптата почки
Заключение: В клубочках и тубулоинтерстициальной системе почки отложений

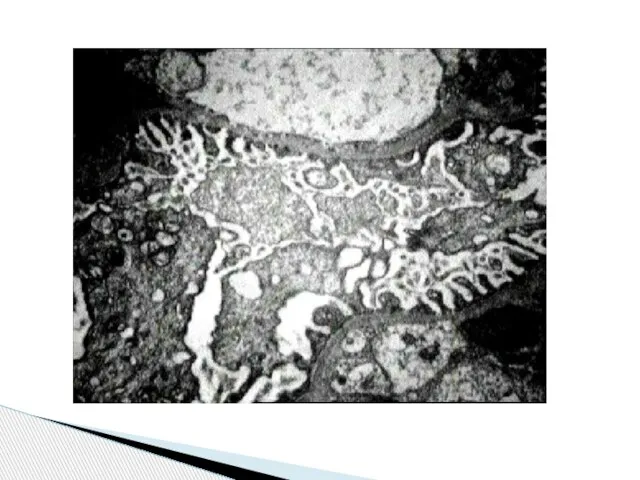
Слайд 83 Болезнь тонкой базальной мембраны с мезангиальной пролиферацией. Сохранная функция почек.
Диагноз:
Болезнь тонкой базальной мембраны с мезангиальной пролиферацией. Сохранная функция почек.
Диагноз:

Слайд 84Не менее 2 больных нефропатией в семье
Гематурия как ведущий симптом нефропатии у
Не менее 2 больных нефропатией в семье
Гематурия как ведущий симптом нефропатии у

Слайд 85Гематурическая форма гломерулонефрита
Болезнь Берже
Дисметаболическая нефропатия
Дифференциальная диагностика
наследственного нефрита
Гематурическая форма гломерулонефрита
Болезнь Берже
Дисметаболическая нефропатия
Дифференциальная диагностика
наследственного нефрита

Слайд 86Генетическое обследование
Полноценное питание
АТФ, кокарбоксилаза, пиридоксин, В15, карнитина хлорид. (Курсы 2 – 3
Генетическое обследование
Полноценное питание
АТФ, кокарбоксилаза, пиридоксин, В15, карнитина хлорид. (Курсы 2 – 3

Слайд 87Нефротический синдром
Липоидный нефроз, или гломерулонефрит с минимальными изменениями
Нефротический синдром
Липоидный нефроз, или гломерулонефрит с минимальными изменениями

Слайд 88Нефротический синдром (НС) -
симптомокомплекс, для которого характерны:
протеинурия у детей не менее 50
Нефротический синдром (НС) -
симптомокомплекс, для которого характерны:
протеинурия у детей не менее 50

Слайд 89В основе НС лежит протеинурия, зависящая от нарушений белков, подоцитов и подоцитарной
В основе НС лежит протеинурия, зависящая от нарушений белков, подоцитов и подоцитарной

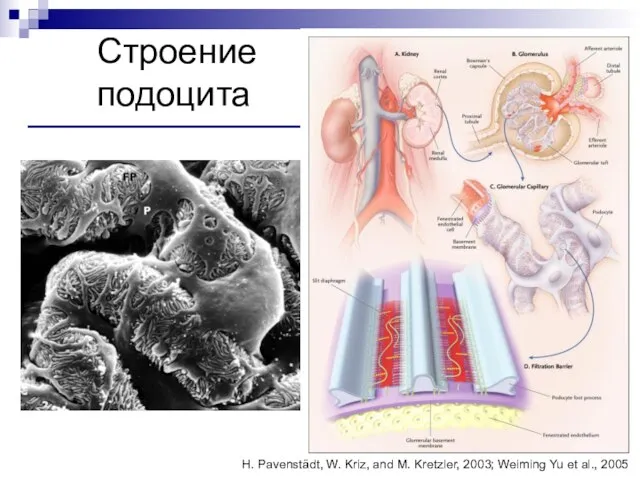
Слайд 90Строение
подоцита
H. Pavenstädt, W. Kriz, and M. Kretzler, 2003; Weiming Yu et al.,
Строение
подоцита
H. Pavenstädt, W. Kriz, and M. Kretzler, 2003; Weiming Yu et al.,
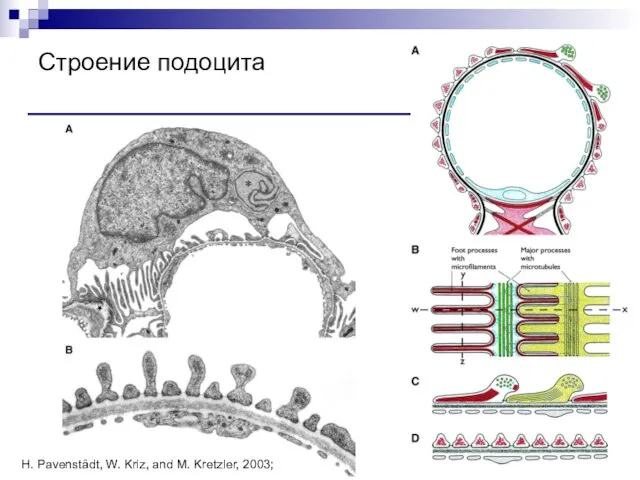
Слайд 91Строение подоцита
H. Pavenstädt, W. Kriz, and M. Kretzler, 2003;
Строение подоцита
H. Pavenstädt, W. Kriz, and M. Kretzler, 2003;
Слайд 92Белки щелевой мембраны
Marilyn G. Farquhar, 2006; H. Pavenstädt, W. Kriz, and M.
Белки щелевой мембраны
Marilyn G. Farquhar, 2006; H. Pavenstädt, W. Kriz, and M.
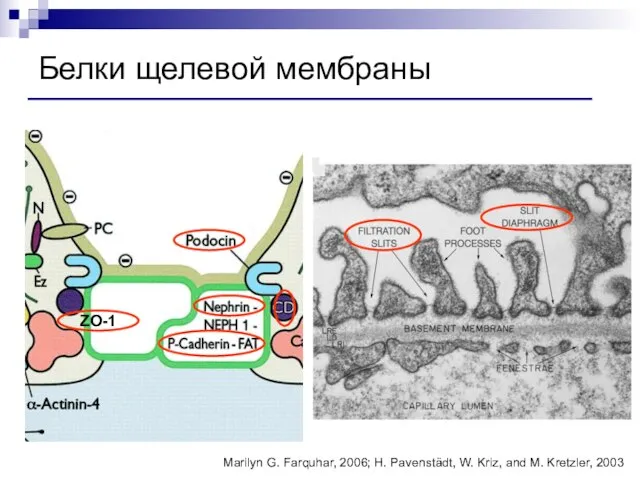
Слайд 93Белки подоцитов и щелевой мембраны
Нефрин
Подоцин
ZO1-мембранный белок
CD2AP (CD2-ассоциированный протеин)
Синаптоподин
Деснин
FAT-трансмембранный адгезивный белок
Белки подоцитов и щелевой мембраны
Нефрин
Подоцин
ZO1-мембранный белок
CD2AP (CD2-ассоциированный протеин)
Синаптоподин
Деснин
FAT-трансмембранный адгезивный белок

Слайд 94Функции подоцитов
Синтез белков гломерулярной базальной мембраны
Регулирование растяжимости клубочкового капилляра
Ограничение попадания в мочевое
Функции подоцитов
Синтез белков гломерулярной базальной мембраны
Регулирование растяжимости клубочкового капилляра
Ограничение попадания в мочевое

Слайд 95В основе НС лежит протеинурия, зависящая от нарушений белков, подоцитов и подоцитарной
В основе НС лежит протеинурия, зависящая от нарушений белков, подоцитов и подоцитарной

Слайд 96Нефротический синдром
Протеинурия – первична
У взрослых – 3,5 г/сутки
У детей:
1г/(м² х
Нефротический синдром
Протеинурия – первична
У взрослых – 3,5 г/сутки
У детей:
1г/(м² х

Слайд 97Нефротический синдром
Гипоальбуминемия при НС 30 – 25 г/л
Нарушения липидного обмена – 3
Нефротический синдром
Гипоальбуминемия при НС 30 – 25 г/л
Нарушения липидного обмена – 3

Слайд 98Нефротический синдром
Отеки в зависимости от внутрисосудистого объема жидкости: гиперволемические,
гиповолемические
нормоволемические
При НС
Нефротический синдром
Отеки в зависимости от внутрисосудистого объема жидкости: гиперволемические,
гиповолемические
нормоволемические
При НС

Слайд 99Врожденный и инфантильный НС
1.Первичный:
Врожденный НС финского типа с микрокистозом
Врожденный НС, французский тип
Врожденный и инфантильный НС
1.Первичный:
Врожденный НС финского типа с микрокистозом
Врожденный НС, французский тип

Слайд 100Врожденный НС финского типа
Finnish Type N.S., finnisher Type NS, неонатальный нефроз
Часто –
Врожденный НС финского типа
Finnish Type N.S., finnisher Type NS, неонатальный нефроз
Часто –

Слайд 101НС с минимальными изменениями –
липоидный нефроз у детей
НС с минимальными измен.
Липоидный
НС с минимальными изменениями –
липоидный нефроз у детей
НС с минимальными измен.
Липоидный

Слайд 102НС с минимальными изменениями –
липоидный нефроз у детей
Возраст – 1,5 –
НС с минимальными изменениями –
липоидный нефроз у детей
Возраст – 1,5 –

Слайд 103Нефротический синдром - определение
НС, преобладающий в структуре НС у детей 1-14 лет,:
Начало
Нефротический синдром - определение
НС, преобладающий в структуре НС у детей 1-14 лет,:
Начало

Слайд 104НС с минимальными изменениями –
липоидный нефроз у детей
Клиника:
отеки при
НС с минимальными изменениями –
липоидный нефроз у детей
Клиника:
отеки при

Слайд 105НС с минимальными изменениями –
липоидный нефроз у детей
Диагноз в 90-95% -
НС с минимальными изменениями –
липоидный нефроз у детей
Диагноз в 90-95% -

Слайд 106Нефротический синдром – варианты течения
Гормоночувствительный НС без рецидивов: нормализация мочи в течение
Нефротический синдром – варианты течения
Гормоночувствительный НС без рецидивов: нормализация мочи в течение

Слайд 107НС с минимальными изменениями –
липоидный нефроз у детей
Течение и исходы:
Острое →
НС с минимальными изменениями –
липоидный нефроз у детей
Течение и исходы:
Острое →

Слайд 108Нефротический синдром – стероидорезистентность
Отсутствие эффекта от преднизолона в максимальной дозе 60 мг/м²/сут
Нефротический синдром – стероидорезистентность
Отсутствие эффекта от преднизолона в максимальной дозе 60 мг/м²/сут

Слайд 109Различия белкового спектра мочи у детей с нефротическим синдромом, в зависимости от
Различия белкового спектра мочи у детей с нефротическим синдромом, в зависимости от
Слайд 110НС с минимальными изменениями –
липоидный нефроз у детей - лечение
Режим: не
НС с минимальными изменениями –
липоидный нефроз у детей - лечение
Режим: не

Слайд 111Нефротический синдром – стероидная зависимость
Рецидив НС при снижении дозы или полной отмене
Нефротический синдром – стероидная зависимость
Рецидив НС при снижении дозы или полной отмене

Слайд 112НС с минимальными изменениями –
липоидный нефроз у детей
Побочные эффекты преднизолона
НС с минимальными изменениями –
липоидный нефроз у детей
Побочные эффекты преднизолона

Слайд 113Нефротический синдром –
показания к цитостатической терапии
Гормонорезистентность в дебюте заболевания
Часто рецидивирующее течение
Развитие
Нефротический синдром –
показания к цитостатической терапии
Гормонорезистентность в дебюте заболевания
Часто рецидивирующее течение
Развитие

Слайд 114НС с минимальными изменениями –
липоидный нефроз у детей - осложнения
Нефротический криз
НС с минимальными изменениями –
липоидный нефроз у детей - осложнения
Нефротический криз

Слайд 115НС с минимальными изменениями –
липоидный нефроз у детей - осложнения
Почечная эклампсия
НС с минимальными изменениями –
липоидный нефроз у детей - осложнения
Почечная эклампсия

Слайд 116Фокально-сегментарный гломерулосклероз
(ФСГС) – диагноз – на данных морфологических исследований
Форма гломерулопатии, для которой
Фокально-сегментарный гломерулосклероз
(ФСГС) – диагноз – на данных морфологических исследований
Форма гломерулопатии, для которой

Слайд 117ФСГС
ФСГС – самая частая причина НС у взрослых (20-25%)
И стероидрезистентного НС
ФСГС
ФСГС – самая частая причина НС у взрослых (20-25%)
И стероидрезистентного НС

Слайд 118ФСГС у детей - лечение
1.После нефробиопсии при стероидрезистентности
2.Детям раннего возраста с субнефротической
ФСГС у детей - лечение
1.После нефробиопсии при стероидрезистентности
2.Детям раннего возраста с субнефротической

Слайд 119НС с минимальными изменениями – цитостатическая терапия
Алкилирующие препараты: (детям не рекомендуются)
Хлорбутин 0,15-0,3мг/кг/сут
НС с минимальными изменениями – цитостатическая терапия
Алкилирующие препараты: (детям не рекомендуются)
Хлорбутин 0,15-0,3мг/кг/сут

 Единая форма размещения информации о проведении диспансеризации. Для сайтов МО
Единая форма размещения информации о проведении диспансеризации. Для сайтов МО Подготовка данных для заполнения формы № 15. Сведения о медицинском наблюдении за состоянием здоровья лиц
Подготовка данных для заполнения формы № 15. Сведения о медицинском наблюдении за состоянием здоровья лиц Анализ работы по выдаче эВСД системы Меркурий
Анализ работы по выдаче эВСД системы Меркурий Анкилостомидозы
Анкилостомидозы Синдромна патологія анемій
Синдромна патологія анемій Стандарты выполнения и оценки результатов исследования внешнего дыхания
Стандарты выполнения и оценки результатов исследования внешнего дыхания Стомотология. Заболевания, связанные с заболеваниями полости рта. Востребованные процедуры
Стомотология. Заболевания, связанные с заболеваниями полости рта. Востребованные процедуры бронхэктаздар
бронхэктаздар Табиғатта таралуы
Табиғатта таралуы Влияние стресса на организм
Влияние стресса на организм Теории питания
Теории питания Триггерные точки по мышцам
Триггерные точки по мышцам Нейровизуализационные методы диагностики заболеваний нервной системы
Нейровизуализационные методы диагностики заболеваний нервной системы Лучевая семиотика при заболеваниях зубочелюстной системы. 2 часть
Лучевая семиотика при заболеваниях зубочелюстной системы. 2 часть Осложнения лекарственной терапии
Осложнения лекарственной терапии Дисгармоничное развитие
Дисгармоничное развитие Средства и методы физической реабилитации при артрите коленного сустава
Средства и методы физической реабилитации при артрите коленного сустава Искусственное прерывание беременности. Имеет ли будущий папа права на принуждение будущей мамы к сохранению беременности
Искусственное прерывание беременности. Имеет ли будущий папа права на принуждение будущей мамы к сохранению беременности НЦД. Болезнь да Коста-Оппенгеймера
НЦД. Болезнь да Коста-Оппенгеймера Введение в урологию. Диагностика урологических заболеваний
Введение в урологию. Диагностика урологических заболеваний Головная боль
Головная боль Патогенез врожденных аномалий женской половой системы
Патогенез врожденных аномалий женской половой системы Онтофилогенетические пороки развития женской половой системы
Онтофилогенетические пороки развития женской половой системы Сыртқы белгілері бойынша адамды сәйкестендіру әдістемесін жетілдіру
Сыртқы белгілері бойынша адамды сәйкестендіру әдістемесін жетілдіру Повреждения органов брюшной полости
Повреждения органов брюшной полости Прямая реставрация передней группы зубов (зубов 1.2-2.2)
Прямая реставрация передней группы зубов (зубов 1.2-2.2) Дренажные дыхательные упражнения. Применение в ЛФК
Дренажные дыхательные упражнения. Применение в ЛФК Схема технологии скрининга на выявление зависимости от психоактивных веществ у подростков
Схема технологии скрининга на выявление зависимости от психоактивных веществ у подростков