- Наследственные синдромы, сопровождающиеся низким ростом

Содержание
- 2. Синдром Ульриха-Нунана
- 3. История: Болезнь описана в 1963 году американским врачом-кардиологом Жаклин Нунан, сообщившей о девяти пациентах со стенозом
- 4. Синдром Нунан представляет собой аутосомно-доминантное заболевание с варьирующей экспрессивностью. Ген синдрома Нунан локализован на длинном плече
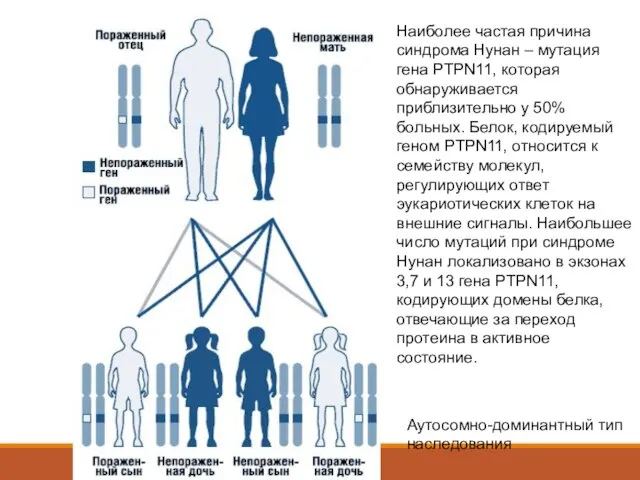
- 5. Аутосомно-доминантный тип наследования Наиболее частая причина синдрома Нунан – мутация гена PTPN11, которая обнаруживается приблизительно у
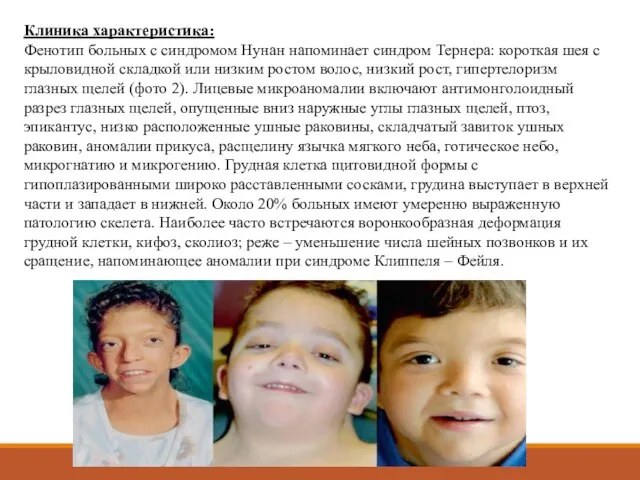
- 6. Клиника характеристика: Фенотип больных с синдромом Нунан напоминает синдром Тернера: короткая шея с крыловидной складкой или
- 7. У больных с синдромом Нунан обычно светлые густые вьющиеся волосы с необычным ростом на темени, часто
- 8. Умственная отсталость выявляется более чем у половины больных, как правило, незначительная. Часто отмечаются особенности поведения, расторможенность,
- 9. Достаточно часто при синдроме Нунан отмечается повышенная кровоточивость, особенно при оперативных вмешательствах в ротовой полости и
- 10. Критерии диагноза Диагноз «синдром Нунан» ставится на основании клинических признаков, в некоторых случаях диагноз подтверждается результатами
- 11. Дифференциальная диагностика У девочек дифференциальный диагноз проводится в первую очередь с синдромом Тернера; уточнить диагноз позволяет
- 12. Представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, необходимость интегрировать отдельные признаки в общий фенотип того или
- 13. Синдром Корнелии де Ланге (синдром Брахмана-Ланге) (амстердамская карликовость)
- 14. Основные проявления болезни в фенотипе -Микроцефалия (уменьшение размеров черепа более чем на 10 % возрастной нормы);
- 15. Встречаемость Частота заболевания — примерно 1 на 10000 ТИП НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ МКБ-10 Q87.1 Синдром Корнелии де
- 16. Основная генетическая причина болезни Примерно половина случаев обусловлена мутациями в гене NIPBL, около 5 % случаев
- 17. У всех больных отмечаются отставание в росте
- 18. Методы диагностики Диагноз синдрома Корнелии де Ланге не всегда прост, так как встречаются умственно отсталые дети
- 19. Лечение Специфического лечения не существует. При необходимости проводят противосудорожную и седативную терапию. Применяют ноотропы, анаболические гормоны
- 20. Синдром Вильямса
- 21. -«лицо эльфа» -тяжелая идиопатическая инфантильная гиперкальциемия -синдром Вильямса -синдром Вильямса - Бойрена -синдром надклапанного стеноза аорты
- 22. синдром Вильямса -генетическое заболевание, в основе которого лежат хромосомные нарушения. -характеризуется особенностями внешнего развития и сопровождается
- 23. причина синдрома Вильямса Делеция (хромосомная перестройка) определённого участка из длинного плеча седьмой хромосомы. Длина потерянного фрагмента
- 24. Где и как часто встречается -встречается в Европе и США -частота неизвестна (равна 1 : 100000
- 25. Гендерный аспект Поражаются оба пола. В литературе есть указание на некоторое преобладание девочек.
- 26. Внешние признаки Эпика́нтус, «монгольская складка» — особая складка у внутреннего угла глаза, в большей или меньшей
- 27. Внешность человека с «синдромом эльфа» в разные возрастные периоды Ребенок 1,5 года Ребенок 6 лет Подросток
- 28. Особенности строения тела -отставание в росте и массе тела -удлиненная шея, узкая грудная клетка, низкая талия,
- 29. Особенности строения тела -Кожа отличается повышенной эластичностью, легко растяжима -На спине, а часто и на щеках
- 30. Особенности психического статуса пациентов с синдромом Вильямса -Нарушения сенсорной интеграции с гиперчувствительностью к звуку и «гравитационной
- 31. Особенности психического статуса пациентов с синдромом Вильямса -Абстрактное мышление у всех больных нарушено очень грубо -Неврозоподобные
- 33. Скачать презентацию

Слайд 3История:
Болезнь описана в 1963 году американским врачом-кардиологом Жаклин Нунан, сообщившей о девяти
История:
Болезнь описана в 1963 году американским врачом-кардиологом Жаклин Нунан, сообщившей о девяти

Слайд 4Синдром Нунан представляет собой аутосомно-доминантное заболевание с варьирующей экспрессивностью. Ген синдрома Нунан
Синдром Нунан представляет собой аутосомно-доминантное заболевание с варьирующей экспрессивностью. Ген синдрома Нунан
Слайд 5Аутосомно-доминантный тип наследования
Наиболее частая причина синдрома Нунан – мутация гена PTPN11,
Аутосомно-доминантный тип наследования
Наиболее частая причина синдрома Нунан – мутация гена PTPN11,
Слайд 6Клиника характеристика:
Фенотип больных с синдромом Нунан напоминает синдром Тернера: короткая шея с
Клиника характеристика:
Фенотип больных с синдромом Нунан напоминает синдром Тернера: короткая шея с
Слайд 7У больных с синдромом Нунан обычно светлые густые вьющиеся волосы с необычным
У больных с синдромом Нунан обычно светлые густые вьющиеся волосы с необычным

Слайд 8Умственная отсталость выявляется более чем у половины больных, как правило, незначительная. Часто
Умственная отсталость выявляется более чем у половины больных, как правило, незначительная. Часто

Слайд 9Достаточно часто при синдроме Нунан отмечается повышенная кровоточивость, особенно при оперативных вмешательствах
Достаточно часто при синдроме Нунан отмечается повышенная кровоточивость, особенно при оперативных вмешательствах

Слайд 10Критерии диагноза
Диагноз «синдром Нунан» ставится на основании клинических признаков, в некоторых случаях
Критерии диагноза
Диагноз «синдром Нунан» ставится на основании клинических признаков, в некоторых случаях

Слайд 11Дифференциальная диагностика
У девочек дифференциальный диагноз проводится в первую очередь с синдромом Тернера;
Дифференциальная диагностика
У девочек дифференциальный диагноз проводится в первую очередь с синдромом Тернера;

Слайд 12Представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, необходимость интегрировать отдельные признаки в
Представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, необходимость интегрировать отдельные признаки в

Слайд 13Синдром Корнелии де Ланге (синдром Брахмана-Ланге) (амстердамская карликовость)
Синдром Корнелии де Ланге (синдром Брахмана-Ланге) (амстердамская карликовость)

Слайд 14Основные проявления болезни в фенотипе
-Микроцефалия (уменьшение размеров черепа более чем на 10 % возрастной
Основные проявления болезни в фенотипе
-Микроцефалия (уменьшение размеров черепа более чем на 10 % возрастной

Слайд 15Встречаемость
Частота заболевания — примерно 1 на 10000
ТИП НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
МКБ-10 Q87.1
Синдром Корнелии де Ланге относится
Встречаемость
Частота заболевания — примерно 1 на 10000
ТИП НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
МКБ-10 Q87.1
Синдром Корнелии де Ланге относится

Слайд 16Основная генетическая причина болезни
Примерно половина случаев обусловлена мутациями в гене NIPBL, около
Основная генетическая причина болезни
Примерно половина случаев обусловлена мутациями в гене NIPBL, около

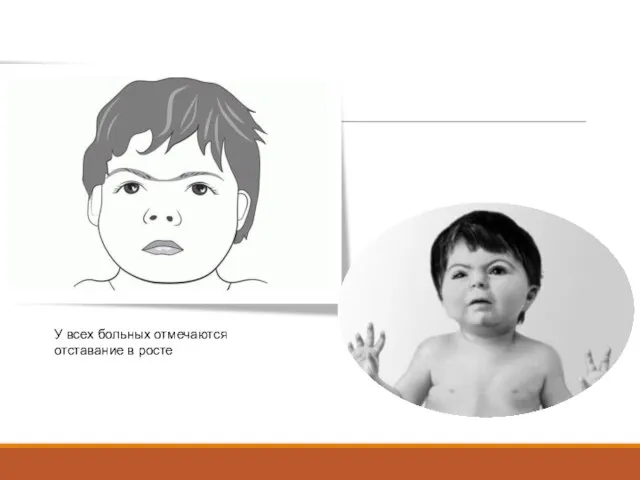
Слайд 17У всех больных отмечаются отставание в росте
У всех больных отмечаются отставание в росте
Слайд 18Методы диагностики
Диагноз синдрома Корнелии де Ланге не всегда прост, так как встречаются
Методы диагностики
Диагноз синдрома Корнелии де Ланге не всегда прост, так как встречаются

Слайд 19Лечение
Специфического лечения не существует. При необходимости проводят противосудорожную и седативную терапию. Применяют
Лечение
Специфического лечения не существует. При необходимости проводят противосудорожную и седативную терапию. Применяют

Слайд 20Синдром Вильямса
Синдром Вильямса

Слайд 21-«лицо эльфа»
-тяжелая идиопатическая инфантильная гиперкальциемия
-синдром Вильямса
-синдром Вильямса - Бойрена
-синдром надклапанного стеноза аорты и умственной отсталости
-«лицо эльфа»
-тяжелая идиопатическая инфантильная гиперкальциемия
-синдром Вильямса
-синдром Вильямса - Бойрена
-синдром надклапанного стеноза аорты и умственной отсталости

Слайд 22синдром Вильямса
-генетическое заболевание, в основе которого лежат хромосомные нарушения.
-характеризуется особенностями внешнего развития
синдром Вильямса
-генетическое заболевание, в основе которого лежат хромосомные нарушения.
-характеризуется особенностями внешнего развития

Слайд 23причина синдрома Вильямса
Делеция (хромосомная перестройка) определённого участка из длинного плеча седьмой
причина синдрома Вильямса
Делеция (хромосомная перестройка) определённого участка из длинного плеча седьмой

Слайд 24Где и как часто встречается
-встречается в Европе и США
-частота неизвестна (равна 1
Где и как часто встречается
-встречается в Европе и США
-частота неизвестна (равна 1

Слайд 25Гендерный аспект
Поражаются оба пола. В литературе есть указание на некоторое преобладание девочек.
Гендерный аспект
Поражаются оба пола. В литературе есть указание на некоторое преобладание девочек.

Слайд 26Внешние признаки
Эпика́нтус, «монгольская складка» — особая складка у внутреннего угла глаза, в большей или
Внешние признаки
Эпика́нтус, «монгольская складка» — особая складка у внутреннего угла глаза, в большей или

Слайд 27Внешность человека с «синдромом эльфа» в разные возрастные периоды
Ребенок 1,5 года
Ребенок 6
Внешность человека с «синдромом эльфа» в разные возрастные периоды
Ребенок 1,5 года
Ребенок 6

Слайд 28Особенности строения тела
-отставание в росте и массе тела
-удлиненная шея, узкая грудная
Особенности строения тела
-отставание в росте и массе тела
-удлиненная шея, узкая грудная

Слайд 29Особенности строения тела
-Кожа отличается повышенной эластичностью, легко растяжима
-На спине, а часто
Особенности строения тела
-Кожа отличается повышенной эластичностью, легко растяжима
-На спине, а часто

Слайд 30Особенности психического статуса пациентов с синдромом Вильямса
-Нарушения сенсорной интеграции с гиперчувствительностью
Особенности психического статуса пациентов с синдромом Вильямса
-Нарушения сенсорной интеграции с гиперчувствительностью

Слайд 31Особенности психического статуса пациентов с синдромом Вильямса
-Абстрактное мышление у всех больных нарушено очень
Особенности психического статуса пациентов с синдромом Вильямса
-Абстрактное мышление у всех больных нарушено очень

 Отчет работа медэксперта
Отчет работа медэксперта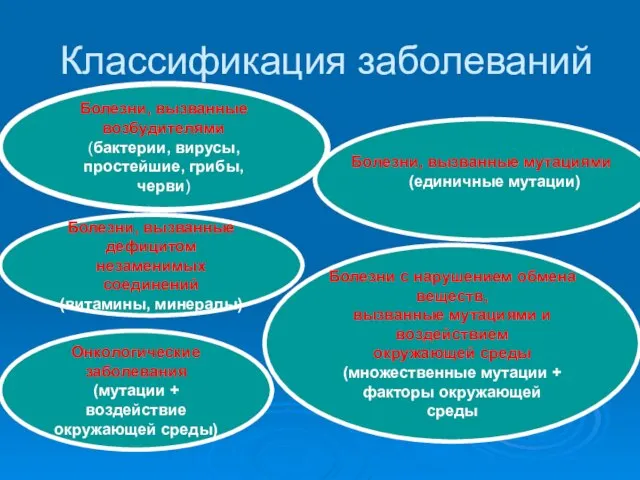 Классификация заболеваний
Классификация заболеваний Планирование беременности как залог здоровья мамы и будущего новорожденного
Планирование беременности как залог здоровья мамы и будущего новорожденного Медицинское страхование 2021-2022
Медицинское страхование 2021-2022 Увеличение продолжительности жизни человека. Методы продления жизни
Увеличение продолжительности жизни человека. Методы продления жизни Клиническая задача. Риск смерти 40 минут
Клиническая задача. Риск смерти 40 минут Клінічні ознаки отруення
Клінічні ознаки отруення Здоровое питание. Понятие
Здоровое питание. Понятие Анафилактический шок
Анафилактический шок Уход за кожей. Сеть клиник Лавиани
Уход за кожей. Сеть клиник Лавиани Глаукома. Что такое глаукома?
Глаукома. Что такое глаукома?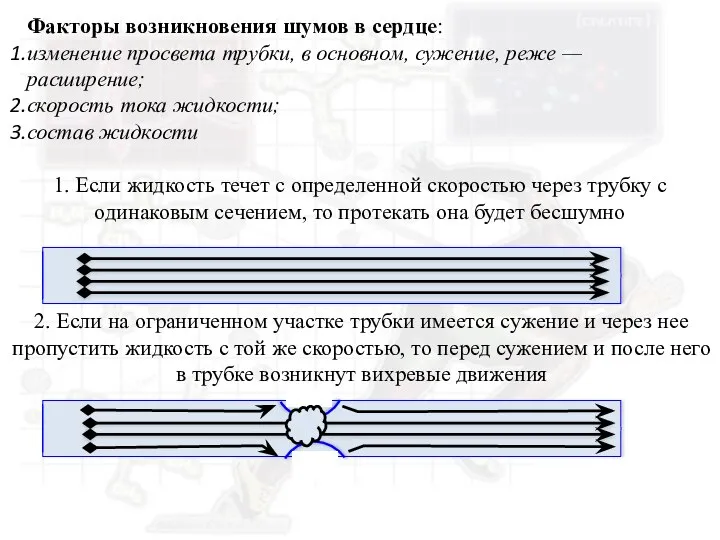 Факторы возникновения шумов в сердце
Факторы возникновения шумов в сердце Здравоохранение Ульяновской области. Национальные проекты
Здравоохранение Ульяновской области. Национальные проекты Җенес генетикасы
Җенес генетикасы Использование мио- и нейростимуляции в спортивной практике
Использование мио- и нейростимуляции в спортивной практике Специальные рентгенологические методы исследования. Ангиография
Специальные рентгенологические методы исследования. Ангиография Лучевая диагностика заболеваний легких
Лучевая диагностика заболеваний легких Сердечно-легочная реанимация
Сердечно-легочная реанимация Бүйрек поликистоз
Бүйрек поликистоз Нейротрансплантация при травмах спинного мозга
Нейротрансплантация при травмах спинного мозга Тромбоэмболия легочной артерии: современные стандарты диагностики, лечения и профилактики. (Новые рекомендации ESC)
Тромбоэмболия легочной артерии: современные стандарты диагностики, лечения и профилактики. (Новые рекомендации ESC) Методики лучевой диагностики
Методики лучевой диагностики Фотометрия. Оптическая схема нефелометра
Фотометрия. Оптическая схема нефелометра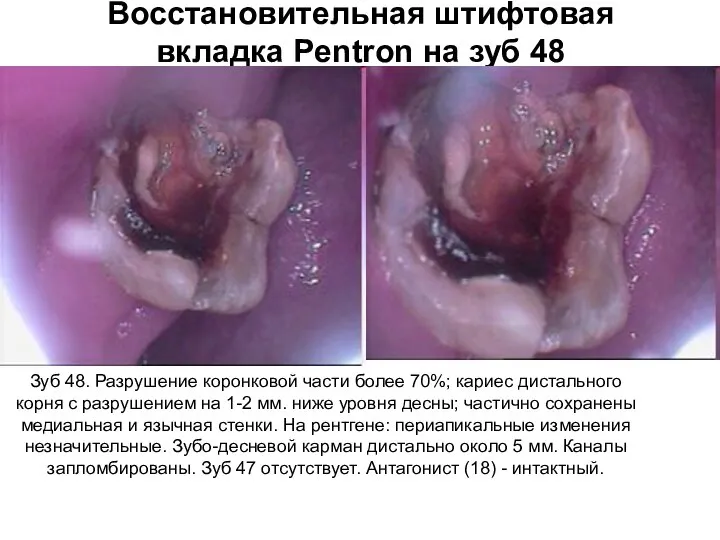 Восстановительная штифтовая вкладка Pentron на зуб 48
Восстановительная штифтовая вкладка Pentron на зуб 48 Сестринское дело в терапии лекция: сестринская помощь при хронической обструктивной болезни легких
Сестринское дело в терапии лекция: сестринская помощь при хронической обструктивной болезни легких Выделение
Выделение Профилактика болезней в пожилом возрасте
Профилактика болезней в пожилом возрасте Рак желудка
Рак желудка