- Синдром Гийен-Барре. Острая воспалительная полирадикулоневропатия аутоиммунной природы

Содержание
- 2. При демиелинизирующем варианте поражаются преимущественно крупные миелинизированные волокна, т. е. двигательные и чувствительные, проводящие глубокую чувствительность.
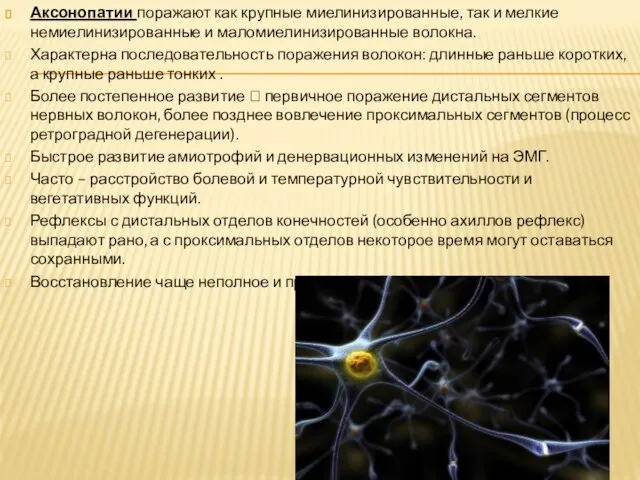
- 3. Аксонопатии поражают как крупные миелинизированные, так и мелкие немиелинизированные и маломиелинизированные волокна. Характерна последовательность поражения волокон:
- 4. Клиническая картина синдрома Гийен-Барре. Прогрессирующий вялый тетрапарез. Вначале слабость чаще вовлекает дистальные или проксимальные отделы ног,
- 5. Более чем у половины больных в остром периоде возникают выраженные вегетативные нарушения: повышение или падение артериального
- 6. Диагноз. Остро или подостро нарастающий вялый тетрапарез ( нижний парапарез), сопровождающийся арефлексией. Характерное течение: прогрессирование в
- 7. Миастения, болезнь Эрба-Гольдфлама аутоиммунное нервно-мышечное заболевание, характеризующееся нарушением нервно-мышечной передачи и проявляющееся слабостью и патологической утомляемостью
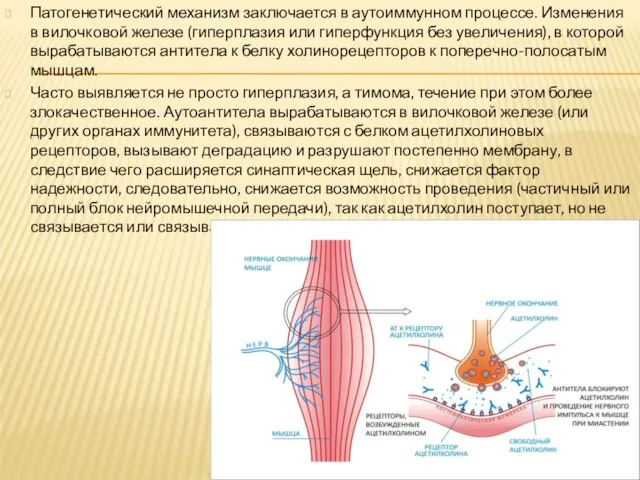
- 8. Патогенетический механизм заключается в аутоиммунном процессе. Изменения в вилочковой железе (гиперплазия или гиперфункция без увеличения), в
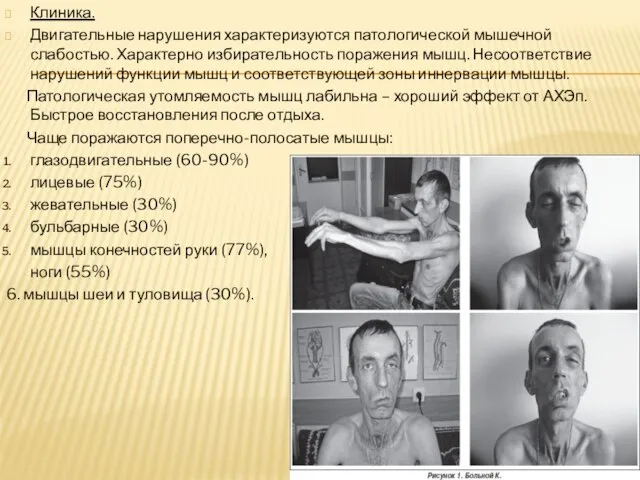
- 9. Клиника. Двигательные нарушения характеризуются патологической мышечной слабостью. Характерно избирательность поражения мышц. Несоответствие нарушений функции мышц и
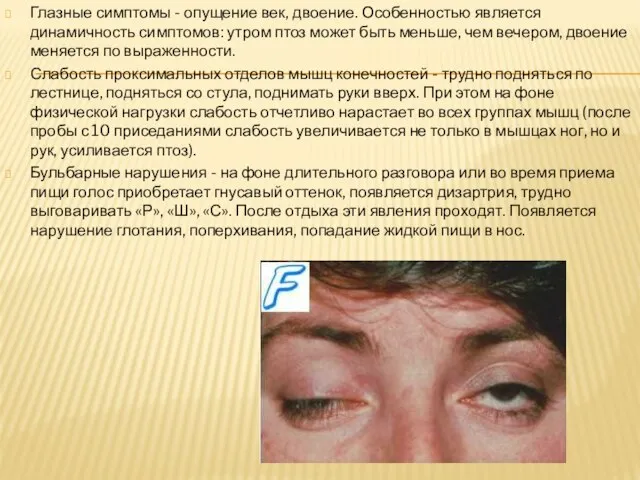
- 10. Глазные симптомы - опущение век, двоение. Особенностью является динамичность симптомов: утром птоз может быть меньше, чем
- 11. 1. Прозериновая проба – вводится Sol.Proserini 0,05% 1-3 мл п/к + Sol.Atropini 0,1% - 0,5 мл
- 12. БАС также известен как болезнь моторных нейронов, Мотонейронная болезнь, болезнь Шарко́, в англоязычных странах — болезнь
- 13. Этиология. Точная этиология БАС неизвестна. Примерно в 5% случаев встречаются семейные (наследственные) формы заболевания. 20% семейных
- 14. Редко наблюдающаяся высокая (церебральная) форма характеризуется спастическим тетрапарезом, псевдобульбарным синдромом при умеренной выраженности признаков повреждения периферического
- 15. Начало БАС с бульбарных нарушений является неблагоприятным прогностическим признаком , поскольку в этих случаях заболевание течет
- 16. По мере прогрессирования заболевания становится невозможным вытягивание губ в трубочку и высасывание языка. Вследствие пареза мышц
- 17. На ногах обычно первыми в процесс вовлекаются передняя и боковая группы мышц, что проявляется “cвисающей стопой
- 18. По мере дегенерации пирамидных путей поверхностные брюшные рефлексы, сохраняющиеся при БАС достаточно длительное время, исчезают. Парестезии
- 19. При БАС обнаружены изменения структуры коллагеновых волокон кожи, чем объясняется парадоксальное отсутствие пролежней у прикованных к
- 20. Выделяют отдельные формы болезни моторного нейрона, являющиеся, вероятно, клинико – морфологическими вариантами БАС. Прогрессирующий бульбарный паралич,
- 21. Первичный боковой склероз - крайне редкое состояние, проявляющееся прогрессирующим нижним спастическим парапарезом с последующим вовлечением верхних
- 22. Диагностика. Всемирная федерация неврологов предлагает следующие критерии диагноза БАС: 1) дегенерация нижнего мотонейрона, доказанная клиническими, электрофизиологическими
- 23. ЭМГ-критерии, подтверждающие диагноз болезни мотонейрона: • фибрилляции и фасцикуляции в мышцах нижних и верхних конечностей, или
- 25. Основой дифференциального диагноза БАС служат следующие признаки: Немиотомное распределение слабости. Слабость у пациентов с БАС обычно
- 26. Полимиозит В 1863 году немецкий врач E. Wagner впервые описал редкое мышечное заболевание, которое он назвал
- 27. Заболевание редкое. По данным Т. A. Medsger и соавт. (1970), болеют 5 человек на 1 млн
- 28. Этиология и патогенез болезни изучены недостаточно. Предполагается персистирующая вирусная инфекция (Коксаки В пикорна-вирусиsдр.). В ряде случаев
- 29. Роль предрасположения в развитии дерматомиозита подтверждается семейной агрегацией аутоиммунных заболеваний, включая дерматомиозит у нескольких членов семьи,
- 30. Классификация, предложенная С. Pearson (1969). Тип I — полимиозит взрослых. Наблюдается главным образом у женщин 30
- 31. КЛАССИФИКАЦИЯ, ПРЕДЛОЖЕННАЯ С. М. BARSON (1966), A. BOHAN И J. В. PETER (1975). В соответствии с
- 32. В РАБОТЕ Е. Л. НАСОНОВА И СОАВТ ( 1995 Г) ВЫДЕЛЕНЫ ФОРМЫ П В ЗАВИСИМОСТИ ОТ
- 33. Согласно классификации Л. В. Догель (1973), дополненной Л. А. Сайковой (1993) выделяют 7 форм хронического полимиозита:
- 34. Одним из наиболее ранних проявлений миозита являются миалгии. Они спонтанные, усиливаются при физической нагрузке и охлаждении.
- 35. Характерный признак – болезненность мышц при пальпации, особенно трапециевидных, дельтовидных, грудных икроножных. Отмечаются боли при растяжении
- 36. Характерны ретракции мышц, возникающие из-за развития миосклероза. Они ограничивают объем пассивных движений и являются отличительной особенностью
- 38. Вследствие слабости мышц тазового пояса изменяется походка, она становится «переваливающейся», миопатической. Кроме скелетной мускулатуры, в патологический
- 39. Поражения кожи разнообразные, они наблюдаются у 35—40% больных, обычно в виде эритемы, развивающейся преимущественно на открытых
- 41. Неврологическая симптоматика представлена поражением центральной и периферической нервной системы. Поражение периферической нервной системы проявляется симптомами поражения
- 42. Полиартралгии (частый признак полимиозита) возникают при движениях и ограничивают подвижность суставов, которая иногда может полностью отсутствовать
- 43. Классическая форма Вагнера-Унферрихта Она объединяет типичные случаи дермато – и полимиозита с острым и подострым течением.
- 45. ПСЕВДОМИОПАТИЧЕСКАЯ ФОРМА Начинается в различном возрасте, нередко после переохлаждения, ОРЗ, ангины гриппа. Развивается постепенно или с
- 47. Псевдоамиотрофическая форма. Форма напоминает наследственные мышечные атрофии ( неврогенные варианты) и включает 2 типа течения: по
- 49. Псевдомиастеническая форма Характерными являются симптомы П. в сочетании с миастеническими явлениями, которые развиваются одновременно или последовательно
- 50. Миосклеротическая форма Редко встречающийся вариант П. заболевание характеризуется массивным развитием миосклероза и контрактур с самого начала
- 51. Миалгическая форма Возникает в возрасте от 15 до 45 лет, чаще у женщин. Основные проявления болезни
- 52. Форма с синдромом Мак-Ардля Характеризуется развитием симптомокомплекса, имеющего большое сходство с гликогенозом 5 типа (болезнью Мак-Ардля),
- 53. Диагностика Лабораторные методы исследования Обязательные: 1) клинический анализ крови (изменения неспецифичны). Увеличение СОЭ (редко – при
- 54. Диагностика Инструментальные методы исследования Обязательные: 1) Электромиография – мышечная возбудимость повышена, потенциалы действия – с низкой
- 55. Диагностические критерии полимиозита (ПМ), предложенные Таnimotо и соавторами (1995): 1) слабость в проксимальных группах мышц верхних,
- 56. Немедикаментозное лечение Лечебная физкультура играет важную роль в предотвращении деформаций. В острой фазе заболевания – ежедневно
- 57. Медикаментозное лечение 1. Глюкокортикостероиды: преднизолон и метилпреднизолон в дозе 1 мг/кг длительно, в среднем, в течение
- 58. 3. В/в иммуноглобулин – по 1 г/кг, в течение 2 дней или по 0,5 г/кг в
- 60. Скачать презентацию
Слайд 2При демиелинизирующем варианте поражаются преимущественно крупные миелинизированные волокна, т. е. двигательные и
При демиелинизирующем варианте поражаются преимущественно крупные миелинизированные волокна, т. е. двигательные и

Слайд 3Аксонопатии поражают как крупные миелинизированные, так и мелкие немиелинизированные и маломиелинизированные волокна.
Характерна
Аксонопатии поражают как крупные миелинизированные, так и мелкие немиелинизированные и маломиелинизированные волокна.
Характерна
Слайд 4Клиническая картина синдрома Гийен-Барре.
Прогрессирующий вялый тетрапарез. Вначале слабость чаще вовлекает дистальные или
Клиническая картина синдрома Гийен-Барре.
Прогрессирующий вялый тетрапарез. Вначале слабость чаще вовлекает дистальные или

Слайд 5Более чем у половины больных в остром периоде возникают выраженные вегетативные нарушения:

Слайд 6Диагноз.
Остро или подостро нарастающий вялый тетрапарез ( нижний парапарез), сопровождающийся арефлексией.
Характерное течение:
Диагноз.
Остро или подостро нарастающий вялый тетрапарез ( нижний парапарез), сопровождающийся арефлексией.
Характерное течение:

Слайд 7Миастения, болезнь Эрба-Гольдфлама
аутоиммунное нервно-мышечное заболевание, характеризующееся нарушением нервно-мышечной передачи и проявляющееся слабостью
Миастения, болезнь Эрба-Гольдфлама
аутоиммунное нервно-мышечное заболевание, характеризующееся нарушением нервно-мышечной передачи и проявляющееся слабостью

Слайд 8Патогенетический механизм заключается в аутоиммунном процессе. Изменения в вилочковой железе (гиперплазия или
Патогенетический механизм заключается в аутоиммунном процессе. Изменения в вилочковой железе (гиперплазия или
Слайд 9Клиника.
Двигательные нарушения характеризуются патологической мышечной слабостью. Характерно избирательность поражения мышц. Несоответствие нарушений
Клиника.
Двигательные нарушения характеризуются патологической мышечной слабостью. Характерно избирательность поражения мышц. Несоответствие нарушений
Слайд 10Глазные симптомы - опущение век, двоение. Особенностью является динамичность симптомов: утром птоз
Глазные симптомы - опущение век, двоение. Особенностью является динамичность симптомов: утром птоз
Слайд 111. Прозериновая проба – вводится
Sol.Proserini 0,05% 1-3 мл п/к +
Sol.Atropini 0,1% -
1. Прозериновая проба – вводится
Sol.Proserini 0,05% 1-3 мл п/к +
Sol.Atropini 0,1% -

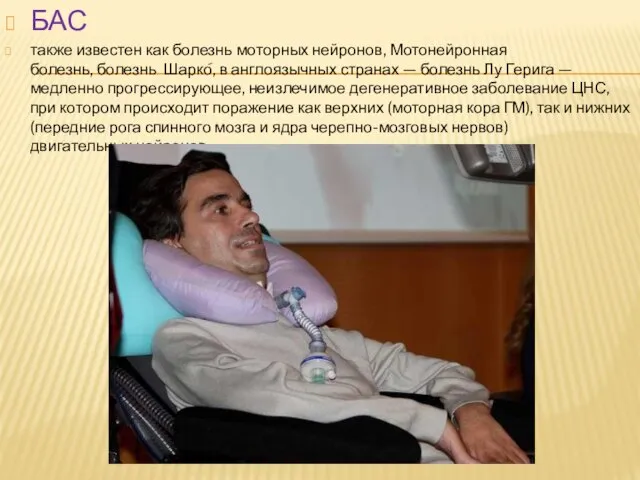
Слайд 12БАС
также известен как болезнь моторных нейронов, Мотонейронная болезнь, болезнь Шарко́, в англоязычных странах — болезнь Лу Герига — медленно
БАС
также известен как болезнь моторных нейронов, Мотонейронная болезнь, болезнь Шарко́, в англоязычных странах — болезнь Лу Герига — медленно
Слайд 13Этиология.
Точная этиология БАС неизвестна. Примерно в 5% случаев встречаются семейные (наследственные)
Этиология.
Точная этиология БАС неизвестна. Примерно в 5% случаев встречаются семейные (наследственные)

Слайд 14Редко наблюдающаяся высокая (церебральная) форма характеризуется спастическим тетрапарезом, псевдобульбарным синдромом при умеренной
Редко наблюдающаяся высокая (церебральная) форма характеризуется спастическим тетрапарезом, псевдобульбарным синдромом при умеренной

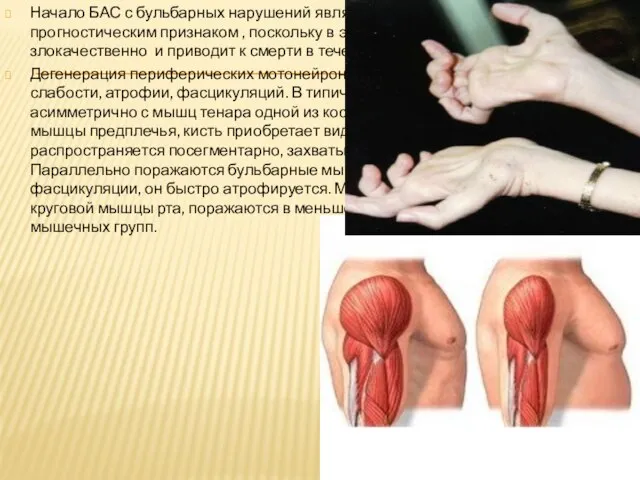
Слайд 15Начало БАС с бульбарных нарушений является неблагоприятным прогностическим признаком , поскольку в
Начало БАС с бульбарных нарушений является неблагоприятным прогностическим признаком , поскольку в
Слайд 16По мере прогрессирования заболевания становится невозможным вытягивание губ в трубочку и высасывание
По мере прогрессирования заболевания становится невозможным вытягивание губ в трубочку и высасывание

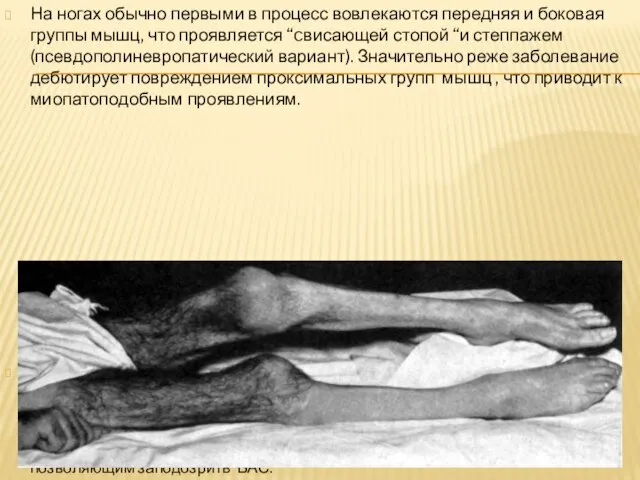
Слайд 17На ногах обычно первыми в процесс вовлекаются передняя и боковая группы мышц,
На ногах обычно первыми в процесс вовлекаются передняя и боковая группы мышц,
Слайд 18По мере дегенерации пирамидных путей поверхностные брюшные рефлексы, сохраняющиеся при БАС достаточно
По мере дегенерации пирамидных путей поверхностные брюшные рефлексы, сохраняющиеся при БАС достаточно

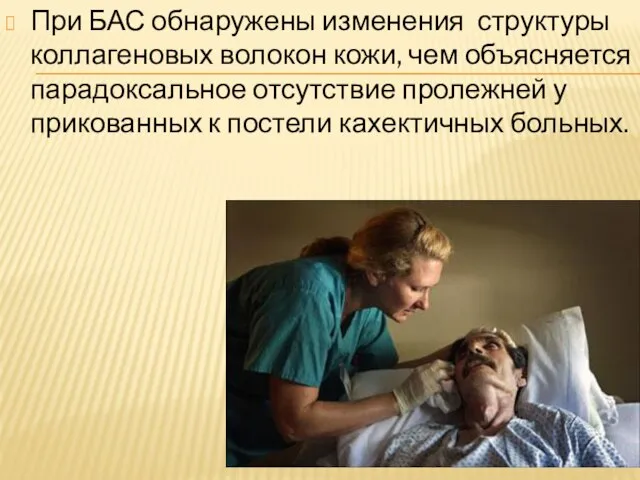
Слайд 19При БАС обнаружены изменения структуры коллагеновых волокон кожи, чем объясняется парадоксальное отсутствие
При БАС обнаружены изменения структуры коллагеновых волокон кожи, чем объясняется парадоксальное отсутствие
Слайд 20Выделяют отдельные формы болезни моторного нейрона, являющиеся, вероятно, клинико – морфологическими вариантами
Выделяют отдельные формы болезни моторного нейрона, являющиеся, вероятно, клинико – морфологическими вариантами

Слайд 21Первичный боковой склероз - крайне редкое состояние, проявляющееся прогрессирующим нижним спастическим парапарезом
Первичный боковой склероз - крайне редкое состояние, проявляющееся прогрессирующим нижним спастическим парапарезом

Слайд 22Диагностика.
Всемирная федерация неврологов предлагает следующие критерии диагноза БАС:
1) дегенерация нижнего мотонейрона, доказанная
Диагностика.
Всемирная федерация неврологов предлагает следующие критерии диагноза БАС:
1) дегенерация нижнего мотонейрона, доказанная

Слайд 23ЭМГ-критерии, подтверждающие диагноз болезни мотонейрона:
• фибрилляции и фасцикуляции в мышцах нижних и верхних конечностей, или
ЭМГ-критерии, подтверждающие диагноз болезни мотонейрона:
• фибрилляции и фасцикуляции в мышцах нижних и верхних конечностей, или

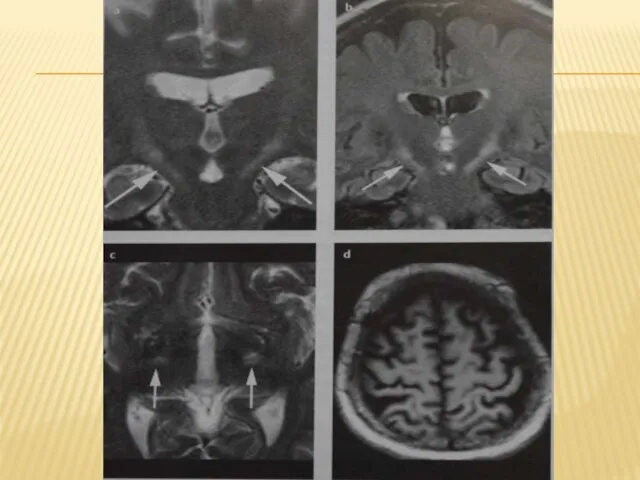
Слайд 25Основой дифференциального диагноза БАС служат следующие признаки:
Немиотомное распределение слабости. Слабость у пациентов
Основой дифференциального диагноза БАС служат следующие признаки:
Немиотомное распределение слабости. Слабость у пациентов

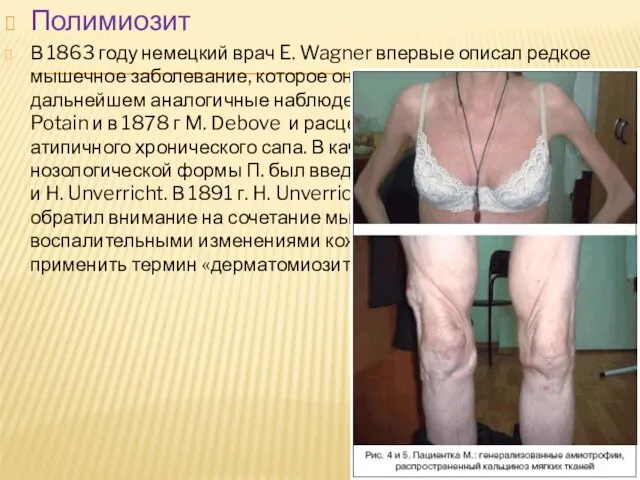
Слайд 26Полимиозит
В 1863 году немецкий врач E. Wagner впервые описал редкое мышечное заболевание,
Полимиозит
В 1863 году немецкий врач E. Wagner впервые описал редкое мышечное заболевание,
Слайд 27Заболевание редкое. По данным Т. A. Medsger и соавт. (1970), болеют 5
Заболевание редкое. По данным Т. A. Medsger и соавт. (1970), болеют 5
Слайд 28Этиология и патогенез болезни изучены недостаточно. Предполагается персистирующая вирусная инфекция (Коксаки В
Этиология и патогенез болезни изучены недостаточно. Предполагается персистирующая вирусная инфекция (Коксаки В
Слайд 29Роль предрасположения в развитии дерматомиозита подтверждается семейной агрегацией аутоиммунных заболеваний, включая дерматомиозит
Роль предрасположения в развитии дерматомиозита подтверждается семейной агрегацией аутоиммунных заболеваний, включая дерматомиозит

Слайд 30Классификация, предложенная С. Pearson (1969).
Тип I — полимиозит взрослых. Наблюдается главным образом у
Классификация, предложенная С. Pearson (1969).
Тип I — полимиозит взрослых. Наблюдается главным образом у

Слайд 31КЛАССИФИКАЦИЯ, ПРЕДЛОЖЕННАЯ С. М. BARSON (1966), A. BOHAN И J. В. PETER
КЛАССИФИКАЦИЯ, ПРЕДЛОЖЕННАЯ С. М. BARSON (1966), A. BOHAN И J. В. PETER

Слайд 32В РАБОТЕ Е. Л. НАСОНОВА И СОАВТ ( 1995 Г) ВЫДЕЛЕНЫ ФОРМЫ
В РАБОТЕ Е. Л. НАСОНОВА И СОАВТ ( 1995 Г) ВЫДЕЛЕНЫ ФОРМЫ
Слайд 33Согласно классификации Л. В. Догель (1973), дополненной Л. А. Сайковой (1993) выделяют
Согласно классификации Л. В. Догель (1973), дополненной Л. А. Сайковой (1993) выделяют

Слайд 34Одним из наиболее ранних проявлений миозита являются миалгии. Они спонтанные, усиливаются при
Одним из наиболее ранних проявлений миозита являются миалгии. Они спонтанные, усиливаются при

Слайд 35Характерный признак – болезненность мышц при пальпации, особенно трапециевидных, дельтовидных, грудных икроножных.
Характерный признак – болезненность мышц при пальпации, особенно трапециевидных, дельтовидных, грудных икроножных.

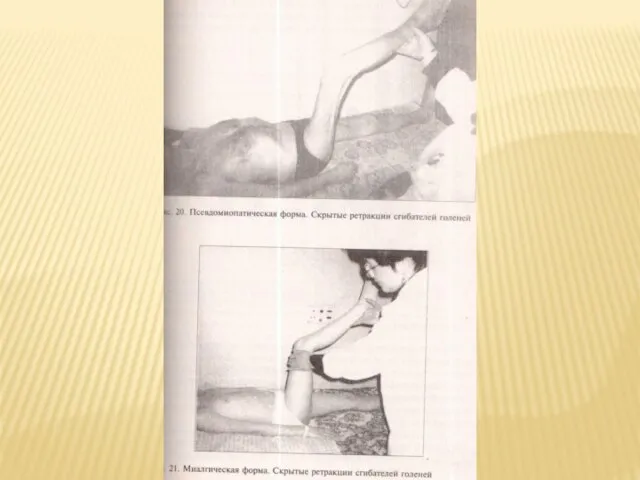
Слайд 36Характерны ретракции мышц, возникающие из-за развития миосклероза. Они ограничивают объем пассивных движений
Характерны ретракции мышц, возникающие из-за развития миосклероза. Они ограничивают объем пассивных движений

Слайд 38Вследствие слабости мышц тазового пояса изменяется походка, она становится «переваливающейся», миопатической.
Кроме скелетной
Вследствие слабости мышц тазового пояса изменяется походка, она становится «переваливающейся», миопатической.
Кроме скелетной

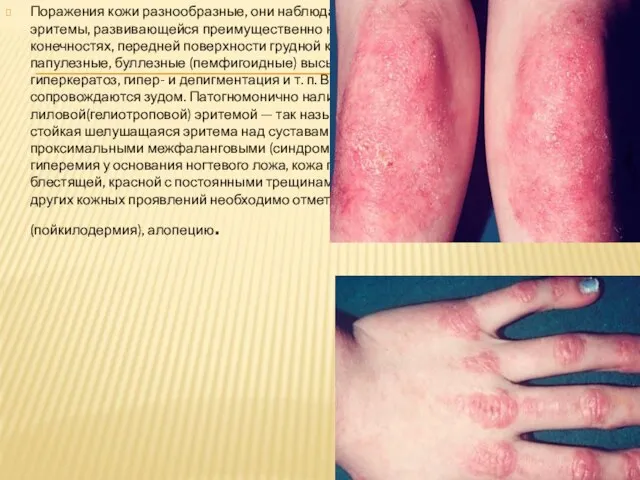
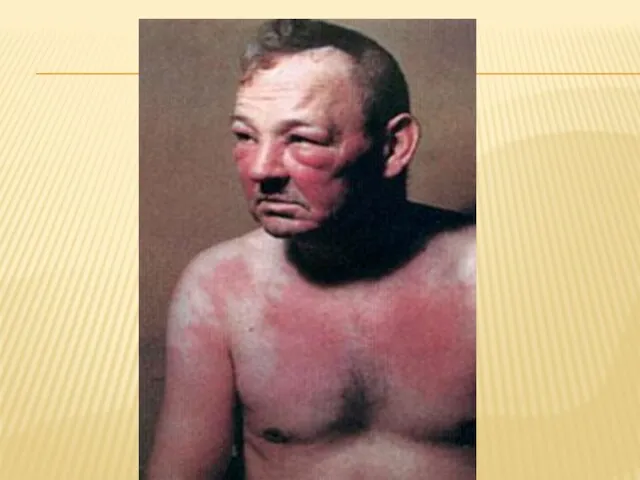
Слайд 39Поражения кожи разнообразные, они наблюдаются у 35—40% больных, обычно в виде эритемы, развивающейся
Поражения кожи разнообразные, они наблюдаются у 35—40% больных, обычно в виде эритемы, развивающейся
Слайд 41Неврологическая симптоматика представлена поражением центральной и периферической нервной системы.
Поражение периферической нервной системы
Неврологическая симптоматика представлена поражением центральной и периферической нервной системы.
Поражение периферической нервной системы

Слайд 42Полиартралгии (частый признак полимиозита) возникают при движениях и ограничивают подвижность суставов, которая
Полиартралгии (частый признак полимиозита) возникают при движениях и ограничивают подвижность суставов, которая

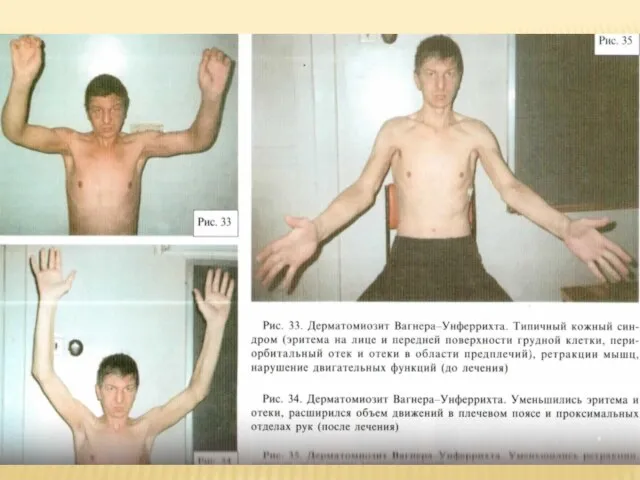
Слайд 43Классическая форма Вагнера-Унферрихта
Она объединяет типичные случаи дермато – и полимиозита с острым
Классическая форма Вагнера-Унферрихта
Она объединяет типичные случаи дермато – и полимиозита с острым

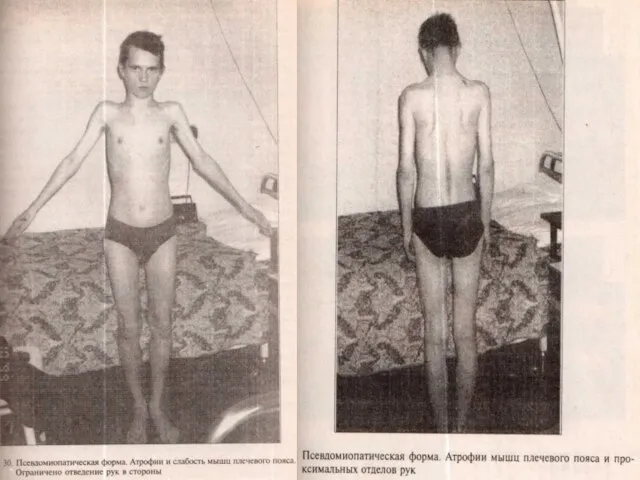
Слайд 45ПСЕВДОМИОПАТИЧЕСКАЯ ФОРМА Начинается в различном возрасте, нередко после переохлаждения, ОРЗ, ангины гриппа.
ПСЕВДОМИОПАТИЧЕСКАЯ ФОРМА Начинается в различном возрасте, нередко после переохлаждения, ОРЗ, ангины гриппа.

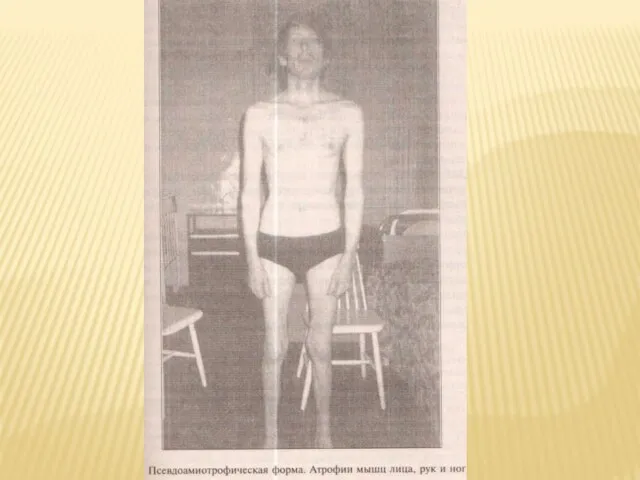
Слайд 47Псевдоамиотрофическая форма.
Форма напоминает наследственные мышечные атрофии ( неврогенные варианты) и включает 2
Псевдоамиотрофическая форма.
Форма напоминает наследственные мышечные атрофии ( неврогенные варианты) и включает 2

Слайд 49Псевдомиастеническая форма
Характерными являются симптомы П. в сочетании с миастеническими явлениями, которые развиваются
Псевдомиастеническая форма
Характерными являются симптомы П. в сочетании с миастеническими явлениями, которые развиваются

Слайд 50Миосклеротическая форма
Редко встречающийся вариант П. заболевание характеризуется массивным развитием миосклероза и контрактур
Миосклеротическая форма
Редко встречающийся вариант П. заболевание характеризуется массивным развитием миосклероза и контрактур

Слайд 51Миалгическая форма
Возникает в возрасте от 15 до 45 лет, чаще у женщин.
Миалгическая форма
Возникает в возрасте от 15 до 45 лет, чаще у женщин.

Слайд 52Форма с синдромом Мак-Ардля
Характеризуется развитием симптомокомплекса, имеющего большое сходство с гликогенозом 5
Форма с синдромом Мак-Ардля
Характеризуется развитием симптомокомплекса, имеющего большое сходство с гликогенозом 5

Слайд 53Диагностика
Лабораторные методы исследования
Обязательные:
1) клинический анализ крови (изменения неспецифичны). Увеличение СОЭ (редко
Диагностика
Лабораторные методы исследования
Обязательные:
1) клинический анализ крови (изменения неспецифичны). Увеличение СОЭ (редко

Слайд 54Диагностика
Инструментальные методы исследования
Обязательные:
1) Электромиография – мышечная возбудимость повышена, потенциалы действия – с
Диагностика
Инструментальные методы исследования
Обязательные:
1) Электромиография – мышечная возбудимость повышена, потенциалы действия – с

Слайд 55Диагностические критерии полимиозита (ПМ), предложенные Таnimotо и соавторами (1995):
1) слабость в проксимальных
Диагностические критерии полимиозита (ПМ), предложенные Таnimotо и соавторами (1995): 1) слабость в проксимальных

Слайд 56Немедикаментозное лечение
Лечебная физкультура играет важную роль в предотвращении деформаций. В острой фазе
Немедикаментозное лечение Лечебная физкультура играет важную роль в предотвращении деформаций. В острой фазе

Слайд 57Медикаментозное лечение
1. Глюкокортикостероиды: преднизолон и метилпреднизолон в дозе 1 мг/кг длительно, в среднем, в
Медикаментозное лечение 1. Глюкокортикостероиды: преднизолон и метилпреднизолон в дозе 1 мг/кг длительно, в среднем, в

Слайд 583. В/в иммуноглобулин – по 1 г/кг, в течение 2 дней или по 0,5
3. В/в иммуноглобулин – по 1 г/кг, в течение 2 дней или по 0,5

 Экстрагенитальные патологии. Острый аппендицит у беременных
Экстрагенитальные патологии. Острый аппендицит у беременных Хирургическая система SmithNephew Werewolf
Хирургическая система SmithNephew Werewolf Влияние токсичных веществ выделяемых из базисов стоматологических протезов, на организм человека
Влияние токсичных веществ выделяемых из базисов стоматологических протезов, на организм человека Бредовые психозы позднего возраста
Бредовые психозы позднего возраста Гнойный средний отит
Гнойный средний отит Жедел көмек көрсету кезінде қажет дәрі – дәрмектерді қолдану
Жедел көмек көрсету кезінде қажет дәрі – дәрмектерді қолдану Жөтел (Ұстама тәрізді жөтел кезіндегі ауруханаға дейінгі шұғыл және жедел көмек көрсету )
Жөтел (Ұстама тәрізді жөтел кезіндегі ауруханаға дейінгі шұғыл және жедел көмек көрсету ) Лечебное дело. Школа биомедицины
Лечебное дело. Школа биомедицины Рецептура. Правила выписывания различных лекарственных форм в рецепте
Рецептура. Правила выписывания различных лекарственных форм в рецепте Студенттердің денсаулығы мен олардың тамақтануы
Студенттердің денсаулығы мен олардың тамақтануы Системный подход к обеспечению сестринского ухода при тромбофлебите нижних конечностей как основа обеспечения качества жизни
Системный подход к обеспечению сестринского ухода при тромбофлебите нижних конечностей как основа обеспечения качества жизни Первая доврачебная помощь при внезапных родах
Первая доврачебная помощь при внезапных родах prezentatsia_po_anatomii_lektsia_1
prezentatsia_po_anatomii_lektsia_1 Аденоидные. Этиология и патогенез
Аденоидные. Этиология и патогенез Общие нарушения жизнедеятельности хирургического больного. Лекция 6
Общие нарушения жизнедеятельности хирургического больного. Лекция 6 Сальмонеллез
Сальмонеллез Муковисцидозбен ауыратын баланың жүрегі мен қан - тамыр жүйесінің функционалдық жағдайы
Муковисцидозбен ауыратын баланың жүрегі мен қан - тамыр жүйесінің функционалдық жағдайы Ангиография. Виды ангиографии
Ангиография. Виды ангиографии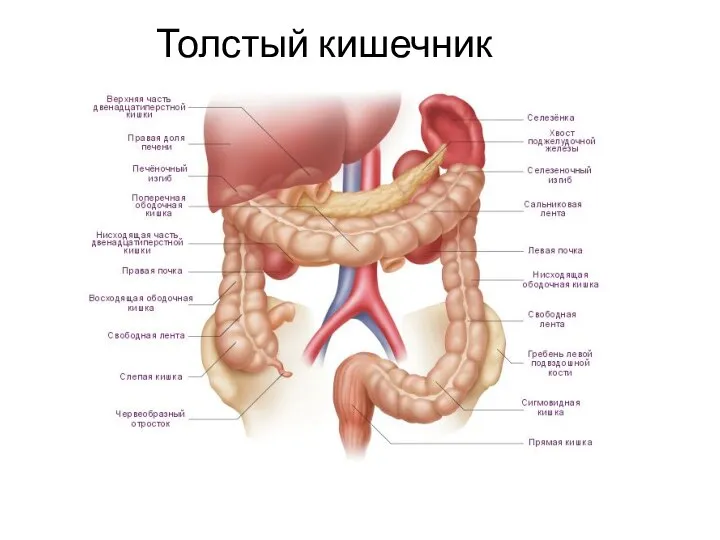 Заболевания и повреждения прямой кишки
Заболевания и повреждения прямой кишки Красный глаз со снижением зрительных функций. Травмы (ранения, контузии, химические, термические и лучевые ожоги)
Красный глаз со снижением зрительных функций. Травмы (ранения, контузии, химические, термические и лучевые ожоги) Анализ ассортимента и наличия лекарственных средств группы макролидов в Республике Беларусь
Анализ ассортимента и наличия лекарственных средств группы макролидов в Республике Беларусь Антибиотиктердің шығу тегіне байланысты түрлері
Антибиотиктердің шығу тегіне байланысты түрлері Плавание и его воздействие на развитие системы опорно-двигательного аппарата
Плавание и его воздействие на развитие системы опорно-двигательного аппарата Формовочные материалы
Формовочные материалы Нефротоксичность аминогликозидных антибиотиков в интенсивной терапии сепсиса
Нефротоксичность аминогликозидных антибиотиков в интенсивной терапии сепсиса Полезно ли мясо для детей и их родителей
Полезно ли мясо для детей и их родителей Диагностика рахита у детей
Диагностика рахита у детей Наследственные заболевания. Расщепление позвоночника
Наследственные заболевания. Расщепление позвоночника