- Болезнь Тея-Сакса

Содержание
- 2. Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) —редкое наследственное заболевание с аутосомно-рецессивным типом
- 3. Тей Уоррен (1843-1927) Сакс Теодор Бернард (1858-1944)
- 4. Гексозаминидаза А (НЕХА) - лизосомный фермент, катализирующий катаболизм GM2 ганглиозида — гексозаминидазы. Он способствует расщеплению жировых
- 5. Различают три формы болезни Тея - Сакса: 1. Детская форма — через полгода после рождения у
- 6. 2. Подростковая форма — развиваются моторно-когнитивные проблемы, дисфагия (нарушение глотания)дизартрия,(расстройства речи), атаксия (шаткость походки), спастичность (контрактуры
- 7. Болезнь распространена у евреев-ашкеназов. Среди них около 3 % являются носителями мутации в гене HEXA. Также
- 8. Клиническая картина: 1. Хронический дефицит гексозаминидазы типа А: Новорожденные в первые месяцы жизни развиваются нормально. Однако,
- 9. 2. Ювенильный дефицит гексозаминидазы типа А. Период начала заболевания с 14 до 30 лет. У взрослых
- 10. Диагностика. Предположительный диагноз ставится после осмотра окулиста. При проверке органов зрения специалист обычно может обнаружить на
- 11. Далее подтвердить предположения помогает анализ на определение количества фермента в жидкостях и тканях исследуемого. Необходимы анализ
- 12. В диагностике заболевания важнейшая роль принадлежит генетическому анализу. Также проводят анализ крови для определения уровня гексозаминидазы
- 13. Определить, есть ли болезнь, до рождения ребенка, позволяет амниоцентез — анализ амниотической жидкости, полученной при проколе
- 15. Болезнь Тея—Сакса не поддается лечению. Клиническая картина обычно нарастает постепенно и также постепенно ведет к угасанию:
- 17. Скачать презентацию
Слайд 2Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) —редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную нервную
Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) —редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную нервную

Слайд 3Тей Уоррен
(1843-1927)
Сакс Теодор Бернард (1858-1944)
Тей Уоррен
(1843-1927)
Сакс Теодор Бернард (1858-1944)

Слайд 4Гексозаминидаза А (НЕХА) - лизосомный фермент, катализирующий катаболизм GM2 ганглиозида — гексозаминидазы.
Гексозаминидаза А (НЕХА) - лизосомный фермент, катализирующий катаболизм GM2 ганглиозида — гексозаминидазы.

Слайд 5Различают три формы болезни
Тея - Сакса:
1. Детская форма — через полгода после
Различают три формы болезни
Тея - Сакса:
1. Детская форма — через полгода после

Слайд 62. Подростковая форма — развиваются моторно-когнитивные проблемы, дисфагия (нарушение глотания)дизартрия,(расстройства речи), атаксия
(шаткость походки), спастичность (контрактуры и параличи). Смерть наступает
2. Подростковая форма — развиваются моторно-когнитивные проблемы, дисфагия (нарушение глотания)дизартрия,(расстройства речи), атаксия
(шаткость походки), спастичность (контрактуры и параличи). Смерть наступает

Слайд 7Болезнь распространена у евреев-ашкеназов. Среди них около 3 % являются носителями мутации в
Болезнь распространена у евреев-ашкеназов. Среди них около 3 % являются носителями мутации в

Слайд 8Клиническая картина:
1. Хронический дефицит гексозаминидазы типа А: Новорожденные в первые месяцы жизни развиваются
Клиническая картина:
1. Хронический дефицит гексозаминидазы типа А: Новорожденные в первые месяцы жизни развиваются

Слайд 92. Ювенильный дефицит гексозаминидазы типа А. Период начала заболевания с 14 до
2. Ювенильный дефицит гексозаминидазы типа А. Период начала заболевания с 14 до

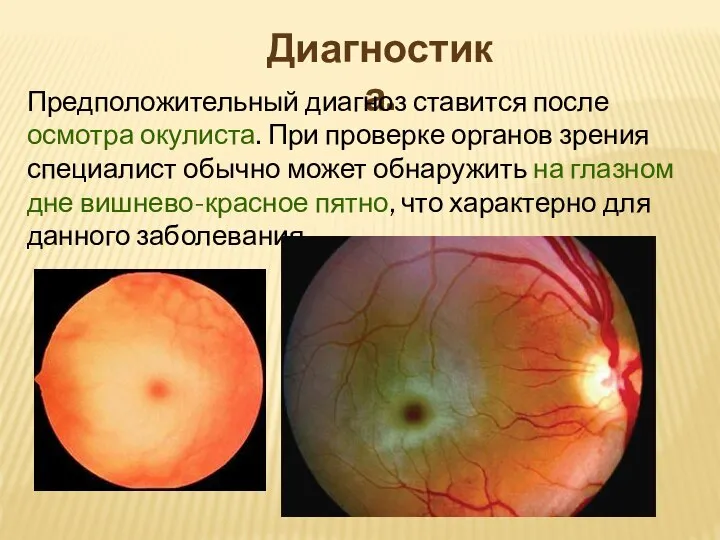
Слайд 10Диагностика.
Предположительный диагноз ставится после осмотра окулиста. При проверке органов зрения специалист
Диагностика.
Предположительный диагноз ставится после осмотра окулиста. При проверке органов зрения специалист
Слайд 11Далее подтвердить предположения помогает анализ на определение количества фермента в жидкостях и
Далее подтвердить предположения помогает анализ на определение количества фермента в жидкостях и
Слайд 12В диагностике заболевания важнейшая роль принадлежит генетическому анализу.
Также проводят анализ крови для
В диагностике заболевания важнейшая роль принадлежит генетическому анализу.
Также проводят анализ крови для

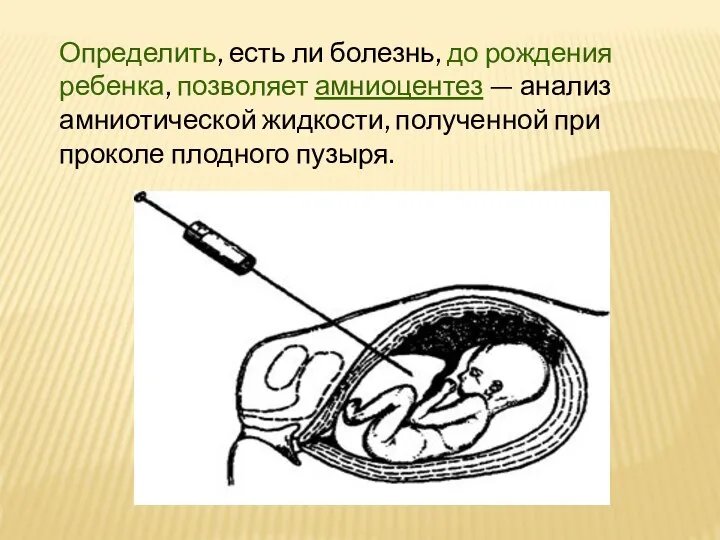
Слайд 13Определить, есть ли болезнь, до рождения ребенка, позволяет амниоцентез — анализ амниотической
Определить, есть ли болезнь, до рождения ребенка, позволяет амниоцентез — анализ амниотической

Слайд 15Болезнь Тея—Сакса не поддается лечению. Клиническая картина обычно нарастает постепенно и также
Болезнь Тея—Сакса не поддается лечению. Клиническая картина обычно нарастает постепенно и также

 Сакроилеит. Факторы
Сакроилеит. Факторы Лук всем детям друг
Лук всем детям друг Основы шва нерва
Основы шва нерва Основы косметических средств фармокологии
Основы косметических средств фармокологии Вторичная атрофия зрительного нерва у детей
Вторичная атрофия зрительного нерва у детей Особенности питания и физических нагрузок при половом созревании у мальчиков
Особенности питания и физических нагрузок при половом созревании у мальчиков Ревматоидный артрит
Ревматоидный артрит Хламидиялық инфекция
Хламидиялық инфекция Гипертрофия небных миндалин
Гипертрофия небных миндалин Новая коронавирусная инфекция у больных хирургического профиля - тактические подходы
Новая коронавирусная инфекция у больных хирургического профиля - тактические подходы Острая почечная недостаточность
Острая почечная недостаточность Денсаулық сақтаудағы қазіргі ақпараттық технология
Денсаулық сақтаудағы қазіргі ақпараттық технология МРТ диагностика компрессионной миелопатии, изменений интра - и эпидуральных пространств
МРТ диагностика компрессионной миелопатии, изменений интра - и эпидуральных пространств Брюшной тиф
Брюшной тиф Инфаркт миокарда. Клинника. Неотложная помощь. Лечение. Реабилитация. Анализ заболеваемости по г. Миасс
Инфаркт миокарда. Клинника. Неотложная помощь. Лечение. Реабилитация. Анализ заболеваемости по г. Миасс Хирургический инструментарий
Хирургический инструментарий Первая помощь при травмах
Первая помощь при травмах Нервная система
Нервная система Основы сосудистого шва
Основы сосудистого шва Трихинеллез животных
Трихинеллез животных Раны
Раны Heat loss from the body
Heat loss from the body Презентация по периоститу
Презентация по периоститу Поступление в организм, транспорт и распределение и выделение ядов. Кумуляция ядов в организме. Коэффициент кумуляции
Поступление в организм, транспорт и распределение и выделение ядов. Кумуляция ядов в организме. Коэффициент кумуляции Лекарство против скуки. Электронная викторина посвященная году медицинского работника
Лекарство против скуки. Электронная викторина посвященная году медицинского работника Иммунодиагностика
Иммунодиагностика Микробиологические основы борьбы с внутрибольничными инфекциями терапевтического отделения
Микробиологические основы борьбы с внутрибольничными инфекциями терапевтического отделения Қатері ісіктердің I-IV кезеңі және TNM бойынша жіктелуі
Қатері ісіктердің I-IV кезеңі және TNM бойынша жіктелуі