- Лизосомы и болезни накопления

Содержание
- 3. Лизосомальные ферменты разрушают макромолекулы либо самой клетки (например, когда перерабатываются структурные компоненты клетки), либо захваченные извне.
- 4. Мукополисахаридозы (МПС) — это группа редких наследственных заболеваний соединительной ткани, связанных с нарушением обмена веществ. Они
- 5. Мукополисахариды содержатся: - на поверхности клеток - во внеклеточном матриксе и его структурах
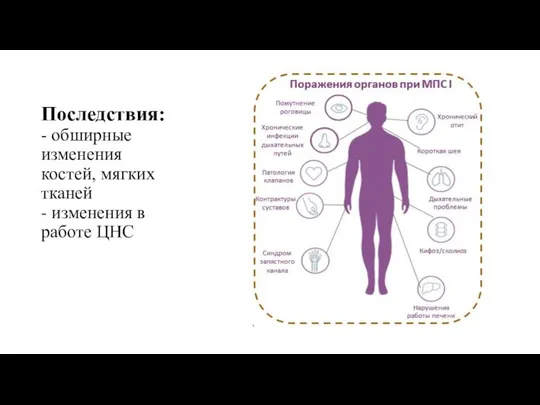
- 6. Последствия: - обширные изменения костей, мягких тканей - изменения в работе ЦНС
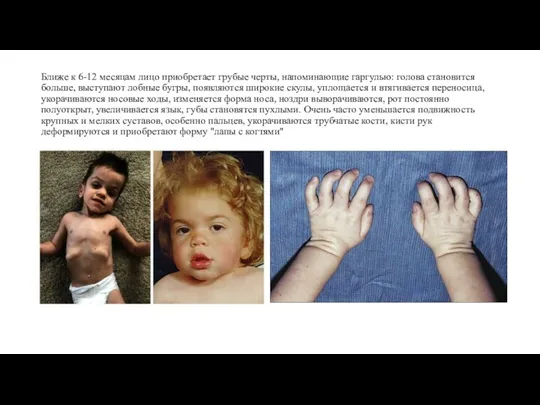
- 7. Ближе к 6-12 месяцам лицо приобретает грубые черты, напоминающие гаргулью: голова становится больше, выступают лобные бугры,
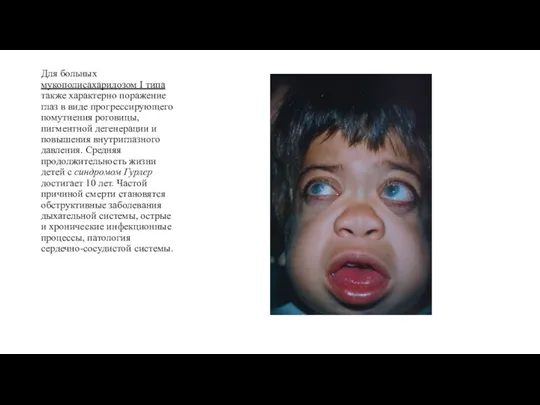
- 8. Для больных мукополисахаридозом I типа также характерно поражение глаз в виде прогрессирующего помутнения роговицы, пигментной дегенерации
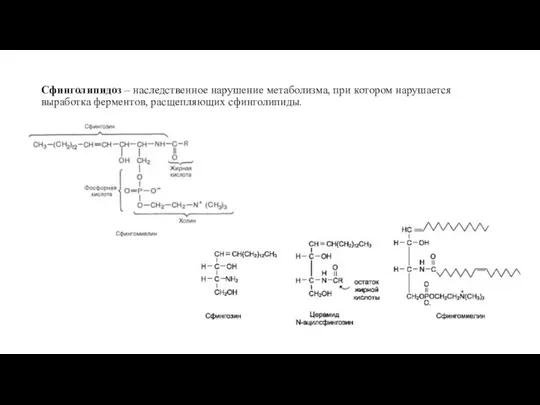
- 9. Сфинголипидоз – наследственное нарушение метаболизма, при котором нарушается выработка ферментов, расщепляющих сфинголипиды.
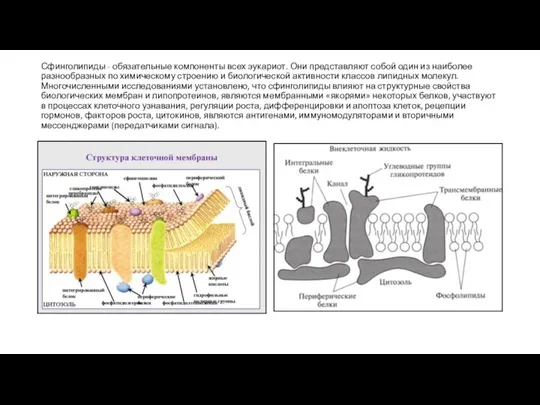
- 10. Сфинголипиды - обязательные компоненты всех эукариот. Они представляют собой один из наиболее разнообразных по химическому строению
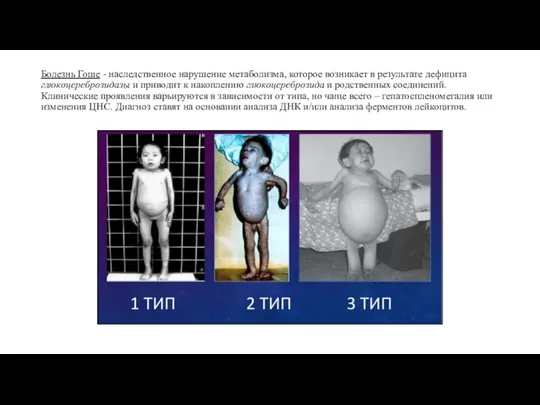
- 11. Болезнь Гоше - наследственное нарушение метаболизма, которое возникает в результате дефицита глюкоцереброзидазы и приводит к накоплению
- 12. Болезнь накопления эфиров холестерола и болезни Вольмана – наследственные нарушения обмена веществ, вызванные дефицитом липазы лизосомальной
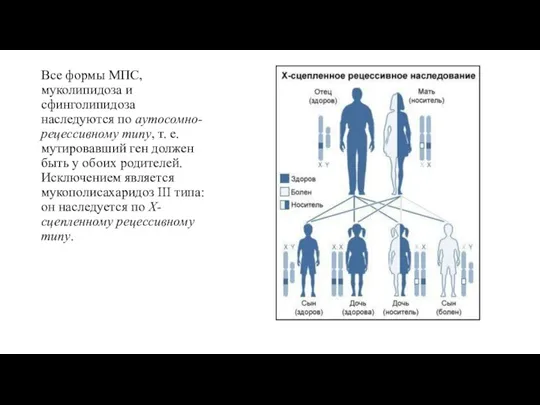
- 13. Все формы МПС, муколипидоза и сфинголипидоза наследуются по аутосомно-рецессивному типу, т. е. мутировавший ген должен быть
- 16. Скачать презентацию

Слайд 3Лизосомальные ферменты разрушают макромолекулы либо самой клетки (например, когда перерабатываются структурные компоненты
Лизосомальные ферменты разрушают макромолекулы либо самой клетки (например, когда перерабатываются структурные компоненты

Слайд 4Мукополисахаридозы (МПС) — это группа редких наследственных заболеваний соединительной ткани, связанных с
Мукополисахаридозы (МПС) — это группа редких наследственных заболеваний соединительной ткани, связанных с

Слайд 5Мукополисахариды содержатся:
- на поверхности клеток
- во внеклеточном матриксе и его структурах
Мукополисахариды содержатся:
- на поверхности клеток
- во внеклеточном матриксе и его структурах

Слайд 6Последствия:
- обширные изменения костей, мягких тканей
- изменения в работе ЦНС
Последствия:
- обширные изменения костей, мягких тканей
- изменения в работе ЦНС
Слайд 7Ближе к 6-12 месяцам лицо приобретает грубые черты, напоминающие гаргулью: голова становится
Ближе к 6-12 месяцам лицо приобретает грубые черты, напоминающие гаргулью: голова становится
Слайд 8Для больных мукополисахаридозом I типа также характерно поражение глаз в виде прогрессирующего
Для больных мукополисахаридозом I типа также характерно поражение глаз в виде прогрессирующего
Слайд 9Сфинголипидоз – наследственное нарушение метаболизма, при котором нарушается выработка ферментов, расщепляющих сфинголипиды.
Сфинголипидоз – наследственное нарушение метаболизма, при котором нарушается выработка ферментов, расщепляющих сфинголипиды.
Слайд 10Сфинголипиды - обязательные компоненты всех эукариот. Они представляют собой один из наиболее
Сфинголипиды - обязательные компоненты всех эукариот. Они представляют собой один из наиболее
Слайд 11Болезнь Гоше - наследственное нарушение метаболизма, которое возникает в результате дефицита глюкоцереброзидазы
Болезнь Гоше - наследственное нарушение метаболизма, которое возникает в результате дефицита глюкоцереброзидазы
Слайд 12Болезнь накопления эфиров холестерола и болезни Вольмана – наследственные нарушения обмена веществ,
Болезнь накопления эфиров холестерола и болезни Вольмана – наследственные нарушения обмена веществ,

Слайд 13Все формы МПС, муколипидоза и сфинголипидоза наследуются по аутосомно-рецессивному типу, т. е.
Все формы МПС, муколипидоза и сфинголипидоза наследуются по аутосомно-рецессивному типу, т. е.
 Нужна помощь
Нужна помощь Эндокринные нарушения
Эндокринные нарушения Антисептики
Антисептики 15 хвилин про цукор
15 хвилин про цукор Efektīvākā terapija
Efektīvākā terapija Осложнения грыж живота
Осложнения грыж живота Внутрисосудистый гемолиз
Внутрисосудистый гемолиз Парентеральды тамақтандыру
Парентеральды тамақтандыру Роды через влагалище, после предыдущего кесарева сечения
Роды через влагалище, после предыдущего кесарева сечения Как попасть в страну здоровья ?
Как попасть в страну здоровья ? Перевод
Перевод Распространенные заболевания ногтей и их предупреждение
Распространенные заболевания ногтей и их предупреждение Аневризмы аорты. Определение
Аневризмы аорты. Определение Экзо- и эндоцервициты
Экзо- и эндоцервициты Черепные нервы
Черепные нервы Правильное питание
Правильное питание Гестозы. Классификация
Гестозы. Классификация История развития проблемы дисфункции носового клапана
История развития проблемы дисфункции носового клапана Профилактика ВБИ
Профилактика ВБИ Секс, половые хромосомы, мужчина, женщина, гемофилия, дальтонизм, сцепленное наследование
Секс, половые хромосомы, мужчина, женщина, гемофилия, дальтонизм, сцепленное наследование Хламидиоз
Хламидиоз Стретчинг в школе
Стретчинг в школе Макроаденома гипофиза. Задача
Макроаденома гипофиза. Задача Язвенный колит(як) и болезнь крона(бк)
Язвенный колит(як) и болезнь крона(бк) Заболевания щитовидной железы
Заболевания щитовидной железы Іс туралы есеп. 17 тістің терең тісжегісі
Іс туралы есеп. 17 тістің терең тісжегісі Применение системы противовирусной защиты для эффективной иммунотерапии рака
Применение системы противовирусной защиты для эффективной иммунотерапии рака Методы диагностики беременности и бесплодия
Методы диагностики беременности и бесплодия