- Наследуемые патологии нарушения обмена углеводов

Содержание
- 2. Наследственные нарушения обмена веществ Наследственные нарушения обмена веществ наследуются аутосомно-рецессивно, аутосомно-доминантно либо рецессивно, сцеплено с полом.
- 3. Стремительный прогресс молекулярной биологии открыл новые возможности диагностики и лечения наследственных нарушений обмена веществ — метод
- 4. Признаки наследственных нарушений обмена веществ Клинические проявления: − тяжелое, угрожающее жизни состояние новорожденного; − задержка физического
- 5. Разные наследственные болезни могут проявляться сходными биохимическими нарушениями. Кроме того, многие клинические проявления наследственных нарушений метаболизма
- 6. При подозрении на наследственное нарушение обмена веществ назначают следующие лабораторные исследования: 1. Общедоступные: − глюкоза в
- 7. Принципы коррекции наследственных нарушений обмена веществ: Коррекция метаболитов: − ограничение субстрата; − восполнение продукта; − направление
- 8. НАРУШЕНИЯ ОБМЕНА ФРУКТОЗЫ У человека известны три наследственных нарушения метаболизма фруктозы: Фруктозурия (недостаточность фруктокиназы) — бессимптомное
- 9. Фруктозурия Этиология. Заболевание наследуется по аутосомнорецессивному типу. Ген кетогексокиназы (КНК) картирован на хромосоме 2р23.3-23.2. Патогенез. Только
- 10. Наследственная непереносимость фруктозы Аутосомно-рецессивное заболевание, обусловленное мутациями гена альдолазы В. Описано около 30 различных мутаций, наиболее
- 11. Наследственная непереносимость фруктозы Диагностика. Наследственная непереносимость фруктозы диагностируется при стандартных биохимических исследованиях — выявляют повышение уровня
- 12. Недостаточность фруктозо-1,6-бифосфатазы Этиология. Аутосомно-рецессивное заболевание, обусловленное мутациями гена фруктозо-1,6- бифосфатазы. Данный ген картирован на хромосоме. Описано
- 13. Недостаточность фруктозо-1,6-бифосфатазы Клиника. Примерно у ½ больных заболевание манифестирует в первые 5 суток жизни гипервентиляционным синдромом
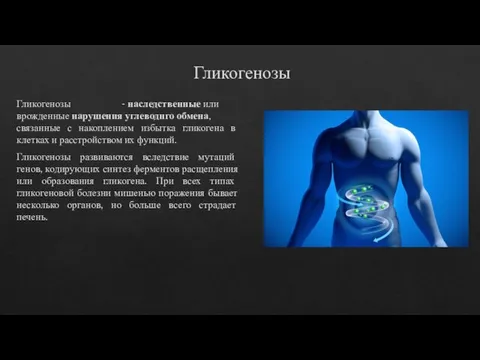
- 14. Гликогенозы Гликогенозы - наследственные или врожденные нарушения углеводнго обмена, связанные с накоплением избытка гликогена в клетках
- 15. Гликогеноз 0 типа (агликеноз) Характеризуется резким снижением запасов гликогена в печени, наблюдается состояние вплоть до развития
- 16. Гликогеноз I типа (болезнь Гирке) Наследуется по аутосомнорецессивному типу и характеризуется нарушением одновременно двух патогенетических процессов:
- 17. Гликогеноз II типа (болезнь Помпе) Наследуется по аутосомнорецессивному типу. Первые симптомы проявляются в первые недели жизни
- 18. Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз) Наследуется по аутосомно-рецессивному типу. Молекула гликогена имеет укороченные
- 19. Особенностью гликогеноза IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени) Является отсутствие повышенного уровня
- 20. Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность) Наследуется по аутосомно-рецессивному типу. Дефект фермента найден в мышцах.
- 21. Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность) Характеризуется образованием гликогена нормальной структуры в достаточном количестве, который
- 22. Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность) По симптоматике сходен с гликогенозом V типа. Дефект фермента
- 23. Гликогеноз VIII типа (болезнь Томпсона) Встречается редко, наследование не установлено. Дефект фермента найден в печени, головном
- 24. Гликогеноз IX типа (болезнь Хага) Наблюдается в виде двух подтипов. Один из них характеризуется аутосомно-рецессивным типом
- 25. Гликогеноз Х типа Самый редкий — известен один случай у единственного больного. Наследование не установлено. Дефект
- 26. Гликогеноз ХI типа характеризуется значительным увеличением печени и резкой задержкой роста. Наблюдаются симптомы гипофосфатемического рахита. Наследование
- 28. МУКОПОЛИСАХАРИДОЗЫ Согласно ферментативным дефектам и тяжести клинической симптоматики, выделяют 10 типов МПС. Фенотипически выделяют следующие типы:
- 29. Мукополисахаридоз
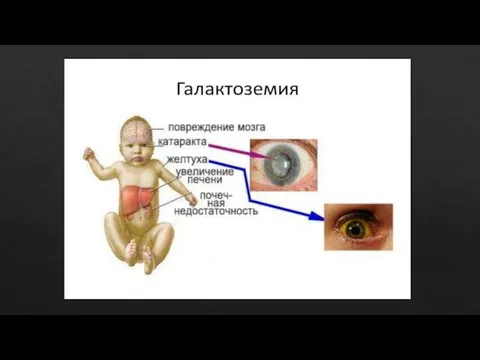
- 30. НАРУШЕНИЯ ОБМЕНА ГАЛАКТОЗЫ Галактоземии— наследственные заболевания, обусловленные нарушениями метаболизма галактозы. Скрининг. Метод для массового неонатального скрининга
- 31. Классификация В настоящее время известно три наследственных заболевания, обусловленных недостаточностью ферментов, участвующих в метаболизме галактозы: −
- 32. Профилактика В мероприятиях по предотвращению галактоземий выделяют два направления. Первичная профилактика: − раннее выявление беременных в
- 35. Скачать презентацию
Слайд 2Наследственные нарушения обмена веществ
Наследственные нарушения обмена веществ наследуются аутосомно-рецессивно, аутосомно-доминантно либо рецессивно,
Наследственные нарушения обмена веществ
Наследственные нарушения обмена веществ наследуются аутосомно-рецессивно, аутосомно-доминантно либо рецессивно,

Слайд 3
Стремительный прогресс молекулярной биологии открыл новые возможности диагностики и лечения
Стремительный прогресс молекулярной биологии открыл новые возможности диагностики и лечения

Слайд 4Признаки наследственных нарушений обмена веществ
Клинические проявления:
− тяжелое, угрожающее жизни состояние новорожденного;
−
Признаки наследственных нарушений обмена веществ
Клинические проявления:
− тяжелое, угрожающее жизни состояние новорожденного;
−

Слайд 5
Разные наследственные болезни могут проявляться сходными биохимическими нарушениями. Кроме того, многие
Разные наследственные болезни могут проявляться сходными биохимическими нарушениями. Кроме того, многие

Слайд 6При подозрении на наследственное нарушение обмена веществ назначают следующие лабораторные исследования:
1.
При подозрении на наследственное нарушение обмена веществ назначают следующие лабораторные исследования:
1.

Слайд 7Принципы коррекции наследственных нарушений обмена веществ:
Коррекция метаболитов:
− ограничение субстрата;
− восполнение
Принципы коррекции наследственных нарушений обмена веществ:
Коррекция метаболитов:
− ограничение субстрата;
− восполнение

Слайд 8НАРУШЕНИЯ ОБМЕНА ФРУКТОЗЫ
У человека известны три наследственных нарушения метаболизма фруктозы:
Фруктозурия (недостаточность
НАРУШЕНИЯ ОБМЕНА ФРУКТОЗЫ
У человека известны три наследственных нарушения метаболизма фруктозы:
Фруктозурия (недостаточность

Слайд 9Фруктозурия
Этиология.
Заболевание наследуется по аутосомнорецессивному типу. Ген кетогексокиназы (КНК) картирован на хромосоме
Фруктозурия
Этиология.
Заболевание наследуется по аутосомнорецессивному типу. Ген кетогексокиназы (КНК) картирован на хромосоме

Слайд 10Наследственная непереносимость фруктозы
Аутосомно-рецессивное заболевание, обусловленное мутациями гена альдолазы В. Описано около 30
Наследственная непереносимость фруктозы
Аутосомно-рецессивное заболевание, обусловленное мутациями гена альдолазы В. Описано около 30

Слайд 11Наследственная непереносимость фруктозы
Диагностика.
Наследственная непереносимость фруктозы диагностируется при стандартных биохимических исследованиях —
Наследственная непереносимость фруктозы
Диагностика.
Наследственная непереносимость фруктозы диагностируется при стандартных биохимических исследованиях —

Слайд 12Недостаточность фруктозо-1,6-бифосфатазы
Этиология.
Аутосомно-рецессивное заболевание, обусловленное мутациями гена фруктозо-1,6- бифосфатазы. Данный ген картирован
Недостаточность фруктозо-1,6-бифосфатазы
Этиология.
Аутосомно-рецессивное заболевание, обусловленное мутациями гена фруктозо-1,6- бифосфатазы. Данный ген картирован

Слайд 13Недостаточность фруктозо-1,6-бифосфатазы
Клиника.
Примерно у ½ больных заболевание манифестирует в первые 5 суток
Недостаточность фруктозо-1,6-бифосфатазы
Клиника.
Примерно у ½ больных заболевание манифестирует в первые 5 суток

Слайд 14 Гликогенозы
Гликогенозы - наследственные или врожденные нарушения углеводнго обмена, связанные с накоплением избытка гликогена в клетках и
Гликогенозы
Гликогенозы - наследственные или врожденные нарушения углеводнго обмена, связанные с накоплением избытка гликогена в клетках и
Слайд 15Гликогеноз 0 типа (агликеноз)
Характеризуется резким снижением запасов гликогена в печени, наблюдается
Гликогеноз 0 типа (агликеноз)
Характеризуется резким снижением запасов гликогена в печени, наблюдается

Слайд 16Гликогеноз I типа (болезнь Гирке)
Наследуется по аутосомнорецессивному типу и характеризуется нарушением
Гликогеноз I типа (болезнь Гирке)
Наследуется по аутосомнорецессивному типу и характеризуется нарушением

Слайд 17Гликогеноз II типа (болезнь Помпе)
Наследуется по аутосомнорецессивному типу.
Первые симптомы проявляются в
Гликогеноз II типа (болезнь Помпе)
Наследуется по аутосомнорецессивному типу.
Первые симптомы проявляются в

Слайд 18Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
Наследуется по аутосомно-рецессивному типу.
Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
Наследуется по аутосомно-рецессивному типу.

Слайд 19Особенностью гликогеноза IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени)
Является
Особенностью гликогеноза IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени)
Является

Слайд 20Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность)
Наследуется по аутосомно-рецессивному типу. Дефект
Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность)
Наследуется по аутосомно-рецессивному типу. Дефект

Слайд 21Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность)
Характеризуется образованием гликогена нормальной структуры в
Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность)
Характеризуется образованием гликогена нормальной структуры в

Слайд 22Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность)
По симптоматике сходен с гликогенозом V
Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность)
По симптоматике сходен с гликогенозом V

Слайд 23Гликогеноз VIII типа (болезнь Томпсона)
Встречается редко, наследование не установлено. Дефект фермента найден
Гликогеноз VIII типа (болезнь Томпсона)
Встречается редко, наследование не установлено. Дефект фермента найден

Слайд 24Гликогеноз IX типа (болезнь Хага)
Наблюдается в виде двух подтипов. Один из них
Гликогеноз IX типа (болезнь Хага)
Наблюдается в виде двух подтипов. Один из них

Слайд 25Гликогеноз Х типа
Самый редкий — известен один случай у единственного больного. Наследование
Гликогеноз Х типа
Самый редкий — известен один случай у единственного больного. Наследование

Слайд 26Гликогеноз ХI типа характеризуется значительным увеличением печени и резкой задержкой роста.
Наблюдаются симптомы
Гликогеноз ХI типа характеризуется значительным увеличением печени и резкой задержкой роста.
Наблюдаются симптомы

Слайд 28МУКОПОЛИСАХАРИДОЗЫ
Согласно ферментативным дефектам и тяжести клинической симптоматики, выделяют 10 типов МПС.
Фенотипически
МУКОПОЛИСАХАРИДОЗЫ
Согласно ферментативным дефектам и тяжести клинической симптоматики, выделяют 10 типов МПС.
Фенотипически

Слайд 29Мукополисахаридоз
Мукополисахаридоз

Слайд 30НАРУШЕНИЯ ОБМЕНА ГАЛАКТОЗЫ
Галактоземии— наследственные заболевания, обусловленные нарушениями метаболизма галактозы.
Скрининг. Метод для массового
НАРУШЕНИЯ ОБМЕНА ГАЛАКТОЗЫ
Галактоземии— наследственные заболевания, обусловленные нарушениями метаболизма галактозы.
Скрининг. Метод для массового

Слайд 31Классификация
В настоящее время известно три наследственных заболевания, обусловленных недостаточностью ферментов, участвующих в
Классификация
В настоящее время известно три наследственных заболевания, обусловленных недостаточностью ферментов, участвующих в

Слайд 32Профилактика
В мероприятиях по предотвращению галактоземий выделяют два направления.
Первичная профилактика:
− раннее
Профилактика
В мероприятиях по предотвращению галактоземий выделяют два направления.
Первичная профилактика:
− раннее

 Гепатопротекторы
Гепатопротекторы Членорасположение плода
Членорасположение плода Алгоритм рациональной дифференциальной диагностики, тактики ведения и динамического наблюдения пациентов при боли в животе
Алгоритм рациональной дифференциальной диагностики, тактики ведения и динамического наблюдения пациентов при боли в животе Фоноритмика
Фоноритмика Витамин Р
Витамин Р Профилактика осложнений ран. Асептика и антисептика
Профилактика осложнений ран. Асептика и антисептика В гепатитінің емі
В гепатитінің емі Дополнительные методы исследования в пульмонологии
Дополнительные методы исследования в пульмонологии Желудочно-кишечные кровотечения
Желудочно-кишечные кровотечения Современные научные исследования в области грудного вскармливания
Современные научные исследования в области грудного вскармливания Нейроонкология. Эпидемиология и статистика
Нейроонкология. Эпидемиология и статистика Альцгеймер
Альцгеймер Осложнения рака ободочной кишки
Осложнения рака ободочной кишки Бета-лактамные антибиотики
Бета-лактамные антибиотики Kurt Julius Isselbacher
Kurt Julius Isselbacher Внутренняя картина болезни: психологические и психические нарушения
Внутренняя картина болезни: психологические и психические нарушения Основные психопатологические синдромы
Основные психопатологические синдромы Общение с пациентами: имеющими различные затруднения (пациенты с культурными, языковыми различиями:)
Общение с пациентами: имеющими различные затруднения (пациенты с культурными, языковыми различиями:) Отравления, помощь
Отравления, помощь Интеллект и интеллектуальная недостаточность. Глубокая умственная отсталость (идиотия)
Интеллект и интеллектуальная недостаточность. Глубокая умственная отсталость (идиотия) Välkommen till kursen Vård och omsorg specialisering, 100 poäng
Välkommen till kursen Vård och omsorg specialisering, 100 poäng Клинические аспекты пневмококковых инфекций
Клинические аспекты пневмококковых инфекций Действие электрического тока на организм человека
Действие электрического тока на организм человека Классификация хирургической инфекции
Классификация хирургической инфекции Методы изучения и гигиеническая оценка комплексного действия метеофакоторов на организм
Методы изучения и гигиеническая оценка комплексного действия метеофакоторов на организм 27.-Dovrachebnaya-pomoshch-krovotechenie-shok-koma
27.-Dovrachebnaya-pomoshch-krovotechenie-shok-koma Стационарзамещающие технологии в онкологической практике
Стационарзамещающие технологии в онкологической практике Методы физиотерапии с заболеваниями ЦНС
Методы физиотерапии с заболеваниями ЦНС