- Пероксисомные болезни

Содержание
- 2. Содержание 1. Определение заболевания 2. Классификация 3. Адренолейкодистрофия а)Клиническая картина б)Лечение в)Диагностика 3. Синдром Целльвегера а)
- 3. Определение заболевания Пероксисомные болезни — гетерогенная группа наследственных болезней обмена веществ, характеризующихся тяжестью течения, выраженным нарушением
- 4. Классификация НАРУШЕНИЯ ДОСТАВКИ БЕЛКОВ В ПЕРОКСИСОМЫ: -синдром Зельвегера -неонатальная адренолейкодистрофия -младенческая болезнь Рефсума -ризомелическая точечная хондродисплазия
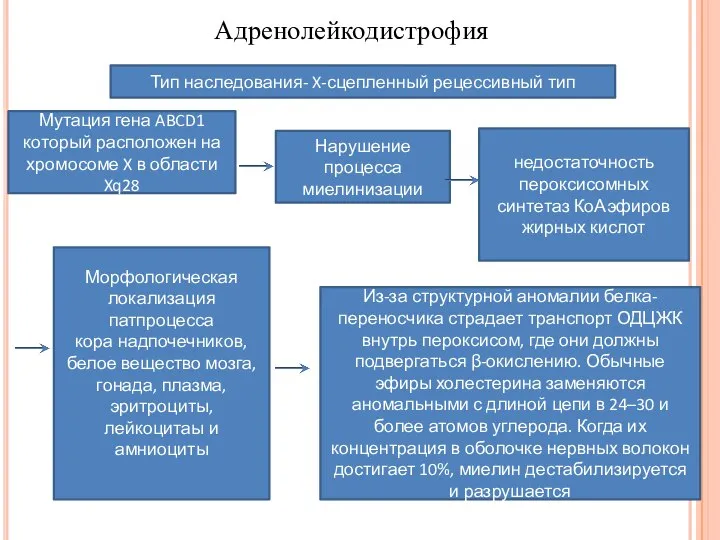
- 5. Адренолейкодистрофия Тип наследования- X-сцепленный рецессивный тип Мутация гена ABCD1 который расположен на хромосоме X в области
- 6. https://bmt.umn.edu/node/120
- 7. Различают шесть клинических форм Х-сцепленной адренолейкодистрофии. Детская церебральная форма Х-сцепленной адренолейкодистрофии. Это наиболее часто наблюдаемая форма
- 8. Взрослая церебральная форма Х-сцепленной адренолейкодистрофии. Заболевание манифестирует после второго десятилетия жизни, клинически сходно с детской церебральной
- 9. Лечение • Неонатальная Методов эффективной теpапии не разработано. Проводится симптоматическая терапия. Необходимо ограничить потребление продуктов с
- 10. Диагностика Наиболее специфичный и информативный лабораторный показатель адренолейкодистрофии — высокая концентрация жирных кислот в плазме, эритроцитах
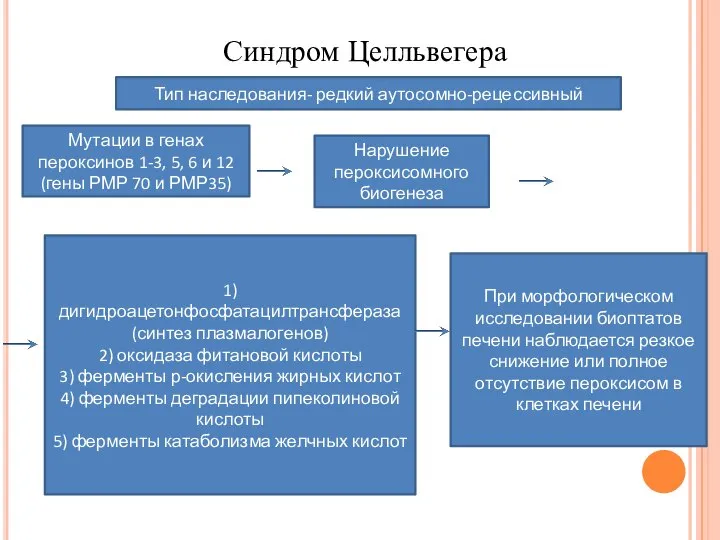
- 11. Синдром Целльвегера Тип наследования- редкий аутосомно-рецессивный Мутации в генах пероксинов 1-3, 5, 6 и 12 (гены
- 12. https://ribem.com.tr/sendromlar/
- 13. Клинические проявления Манифистация в раннем неонатальном периоде. Задержка физического и умственного развития, множественные врожденные дефекты (например,
- 14. Лечение К сожалению, лекарств от синдрома Цельвегера на сегодняшний день не существует. Можно использовать некоторые медицинские
- 15. Диагностика При исследовании крови определяются повышение жирных кислот с очень длинной углеродной цепью. Определение активности дигидроацетон-фос-
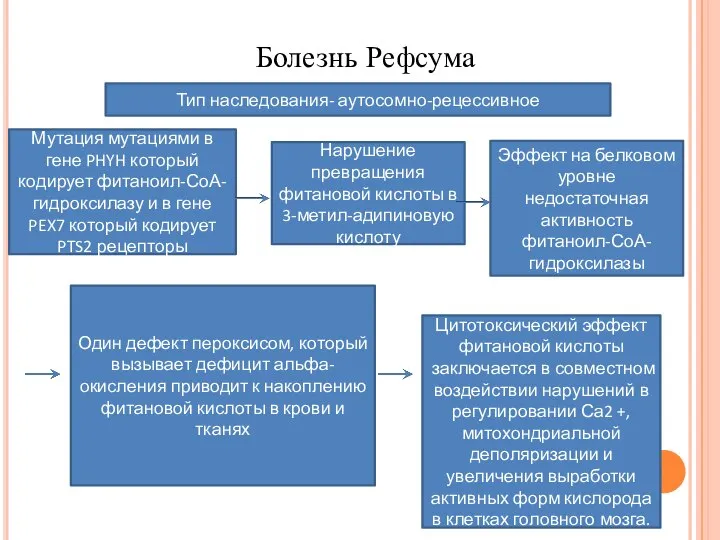
- 16. Болезнь Рефсума Тип наследования- аутосомно-рецессивное Мутация мутациями в гене PHYH который кодирует фитаноил-СоА-гидроксилазу и в гене
- 17. https://www.omicsonline.org/singapore/charcotmarietooth-disease-peer-reviewed-pdf-ppt-articles/
- 18. Клиническая картина Наблюдаются в подростковом возрасте и характеризуются поражением периферической нервной системы (полиневропатия), мозжечковой атаксией, нарушением
- 19. Лечение • Медикаментозного лечения не существует. Лечение симптоматическое. Катаракты необходимо удалять в раннем возрасте, слуховые аппараты,замещение
- 20. Диагностика • Биохимический анализ крови Нарушение метаболизма пероксисомальных ЖК выявляют по уровню длинноцепочечных жирных кислот в
- 22. Скачать презентацию
Слайд 2Содержание
1. Определение заболевания
2. Классификация
3. Адренолейкодистрофия
а)Клиническая картина
б)Лечение
в)Диагностика
3. Синдром Целльвегера
а) Клиническая
Содержание
1. Определение заболевания
2. Классификация
3. Адренолейкодистрофия
а)Клиническая картина
б)Лечение
в)Диагностика
3. Синдром Целльвегера
а) Клиническая

Слайд 3Определение заболевания
Пероксисомные болезни — гетерогенная группа наследственных болезней обмена веществ, характеризующихся тяжестью
Определение заболевания
Пероксисомные болезни — гетерогенная группа наследственных болезней обмена веществ, характеризующихся тяжестью

Слайд 4Классификация
НАРУШЕНИЯ ДОСТАВКИ БЕЛКОВ В ПЕРОКСИСОМЫ:
-синдром Зельвегера
-неонатальная адренолейкодистрофия
-младенческая болезнь Рефсума
-ризомелическая точечная
Классификация
НАРУШЕНИЯ ДОСТАВКИ БЕЛКОВ В ПЕРОКСИСОМЫ:
-синдром Зельвегера
-неонатальная адренолейкодистрофия
-младенческая болезнь Рефсума
-ризомелическая точечная

Слайд 5Адренолейкодистрофия
Тип наследования- X-сцепленный рецессивный тип
Мутация гена ABCD1 который расположен на хромосоме
Адренолейкодистрофия
Тип наследования- X-сцепленный рецессивный тип
Мутация гена ABCD1 который расположен на хромосоме
Слайд 6https://bmt.umn.edu/node/120
https://bmt.umn.edu/node/120

Слайд 7Различают шесть клинических форм Х-сцепленной адренолейкодистрофии.
Детская церебральная форма Х-сцепленной адренолейкодистрофии. Это наиболее
Различают шесть клинических форм Х-сцепленной адренолейкодистрофии.
Детская церебральная форма Х-сцепленной адренолейкодистрофии. Это наиболее

Слайд 8Взрослая церебральная форма Х-сцепленной адренолейкодистрофии. Заболевание манифестирует после второго десятилетия жизни, клинически
Взрослая церебральная форма Х-сцепленной адренолейкодистрофии. Заболевание манифестирует после второго десятилетия жизни, клинически

Слайд 9Лечение
• Неонатальная
Методов эффективной теpапии не разработано. Проводится симптоматическая терапия. Необходимо ограничить потребление
Лечение
• Неонатальная
Методов эффективной теpапии не разработано. Проводится симптоматическая терапия. Необходимо ограничить потребление

Слайд 10Диагностика
Наиболее специфичный и информативный лабораторный показатель адренолейкодистрофии — высокая концентрация жирных кислот
Диагностика
Наиболее специфичный и информативный лабораторный показатель адренолейкодистрофии — высокая концентрация жирных кислот

Слайд 11Синдром Целльвегера
Тип наследования- редкий аутосомно-рецессивный
Мутации в генах пероксинов 1-3, 5, 6 и
Синдром Целльвегера
Тип наследования- редкий аутосомно-рецессивный
Мутации в генах пероксинов 1-3, 5, 6 и
Слайд 12https://ribem.com.tr/sendromlar/
https://ribem.com.tr/sendromlar/
Слайд 13Клинические проявления
Манифистация в раннем неонатальном периоде. Задержка физического и умственного развития, множественные
Клинические проявления
Манифистация в раннем неонатальном периоде. Задержка физического и умственного развития, множественные

Слайд 14Лечение
К сожалению, лекарств от синдрома Цельвегера на сегодняшний день не существует. Можно
Лечение
К сожалению, лекарств от синдрома Цельвегера на сегодняшний день не существует. Можно

Слайд 15Диагностика
При исследовании крови определяются повышение жирных кислот с очень длинной углеродной цепью.
Диагностика
При исследовании крови определяются повышение жирных кислот с очень длинной углеродной цепью.

Слайд 16Болезнь Рефсума
Тип наследования- аутосомно-рецессивное
Мутация мутациями в гене PHYH который кодирует фитаноил-СоА-гидроксилазу
Болезнь Рефсума
Тип наследования- аутосомно-рецессивное
Мутация мутациями в гене PHYH который кодирует фитаноил-СоА-гидроксилазу
Слайд 17https://www.omicsonline.org/singapore/charcotmarietooth-disease-peer-reviewed-pdf-ppt-articles/
https://www.omicsonline.org/singapore/charcotmarietooth-disease-peer-reviewed-pdf-ppt-articles/
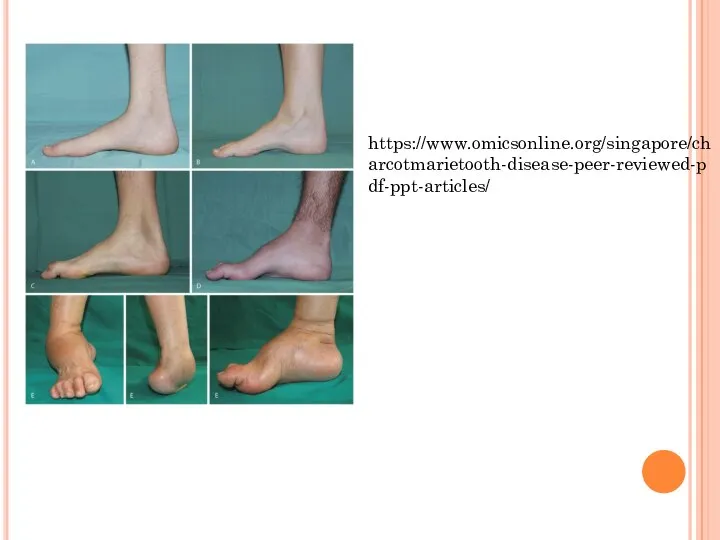
Слайд 18Клиническая картина
Наблюдаются в подростковом возрасте и характеризуются поражением периферической нервной системы (полиневропатия),
Клиническая картина
Наблюдаются в подростковом возрасте и характеризуются поражением периферической нервной системы (полиневропатия),

Слайд 19Лечение
• Медикаментозного лечения не существует. Лечение симптоматическое. Катаракты необходимо удалять в раннем
Лечение
• Медикаментозного лечения не существует. Лечение симптоматическое. Катаракты необходимо удалять в раннем

Слайд 20Диагностика
• Биохимический анализ крови
Нарушение метаболизма пероксисомальных ЖК выявляют по уровню длинноцепочечных жирных
Диагностика
• Биохимический анализ крови
Нарушение метаболизма пероксисомальных ЖК выявляют по уровню длинноцепочечных жирных

 Метод ПЦР в диагностике инфекционных заболеваний
Метод ПЦР в диагностике инфекционных заболеваний Дифференциальная диагностика при мочевом синдроме
Дифференциальная диагностика при мочевом синдроме Биомикроскопия глаза
Биомикроскопия глаза ТОО Прикасйпийский региональный центр охраны материнства и детства и экотоксикологии Caspian Clinic
ТОО Прикасйпийский региональный центр охраны материнства и детства и экотоксикологии Caspian Clinic Информация к размышлению
Информация к размышлению Биопсия при проведении УЗИ
Биопсия при проведении УЗИ Некробактеріоз. Визначення хвороби
Некробактеріоз. Визначення хвороби Правильное питание
Правильное питание Структура Саратовского МНЦ гигиены ФБУН ФНЦ медико-профилактических технологий управления рисками здоровью населения
Структура Саратовского МНЦ гигиены ФБУН ФНЦ медико-профилактических технологий управления рисками здоровью населения Влияние системных заболеваний соединительной ткани на состояние пародонта
Влияние системных заболеваний соединительной ткани на состояние пародонта Уход и наблюдение за детьми грудного возраста при заболеваниях органов дыхания
Уход и наблюдение за детьми грудного возраста при заболеваниях органов дыхания Балапандардың жынысын тестілеу үшін арналған құрылғыны дамыту
Балапандардың жынысын тестілеу үшін арналған құрылғыны дамыту Стационарлардағы медициналық көмектің сапасын қамтамасыз етудегі дәрілік формулярлар жүйесі
Стационарлардағы медициналық көмектің сапасын қамтамасыз етудегі дәрілік формулярлар жүйесі Подростковый наркотизм и другие аддикции
Подростковый наркотизм и другие аддикции Эндовидеохирургические технологии в лечении грыж
Эндовидеохирургические технологии в лечении грыж Требования к ситуационной задаче
Требования к ситуационной задаче Подагра
Подагра Неотложные состояния в неонатологии
Неотложные состояния в неонатологии Сон в короне
Сон в короне Оказание первой помощи детям при несчастных случаях, травмах, отравлениях и состояниях, угрожающих жизни и здоровью
Оказание первой помощи детям при несчастных случаях, травмах, отравлениях и состояниях, угрожающих жизни и здоровью Микробиологические основы химиотерапии и химиопрофилактики инфекционных заболеваний
Микробиологические основы химиотерапии и химиопрофилактики инфекционных заболеваний Расстройства личности
Расстройства личности День борьбы с артритом
День борьбы с артритом Опухоли носа и околоносовых пазух
Опухоли носа и околоносовых пазух Электронные микрофотографии SARS-COV-2
Электронные микрофотографии SARS-COV-2 Болезни передаваемые половым путем
Болезни передаваемые половым путем Упражнения для укрепления мышц
Упражнения для укрепления мышц Реабилитация при сердечно-сосудистых заболеваниях
Реабилитация при сердечно-сосудистых заболеваниях