Слайд 2Определение
-это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур.

Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.
Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин "краниофациальная микросомия". Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Слайд 4Этиология
Изучена недостаточно, возможно – аутосомно-доминантный. Ген, детерминирующий клинические проявления синдрома

Гольденхара, картирован на длинном плече хромосомы 14, в локусе 14q32. У пациентов с фенотипом синдрома Гольденхара могут встречаться также разные хромосомные аномалии.
Большинство случаев синдрома - спорадические. Также играют роль неблагоприятный акушерско-гинекологический анамнез матери (предшествующие аборты, сахарный диабет, избыточный вес) и тератогенные факторы на ранних сроках беременности.
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
приём некоторых лекарственных препаратов, противопоказанных при беременности;
вредные привычки;
химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития.
Слайд 5Клинические проявления
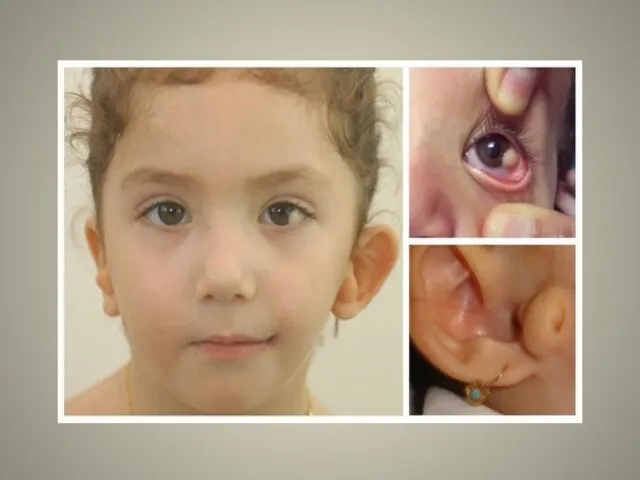
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти)
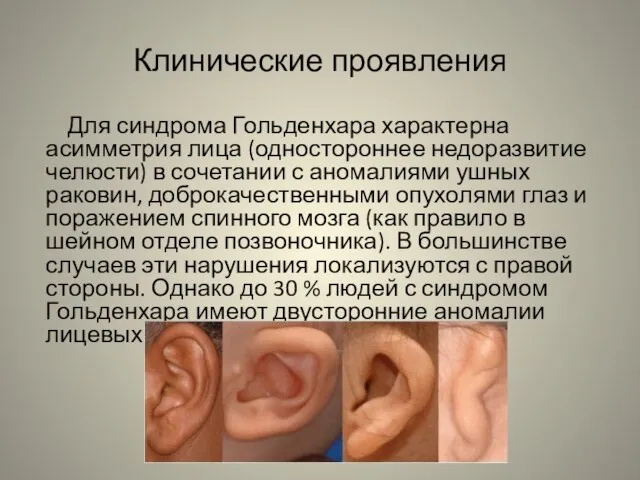
в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны. Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
Слайд 6Клинические проявления
К лицевым аномалиям синдрома относятся:
расщелины лица и неба, аномалии лицевых

мышц, верхней и нижней челюстей, скуловой и височной костей;
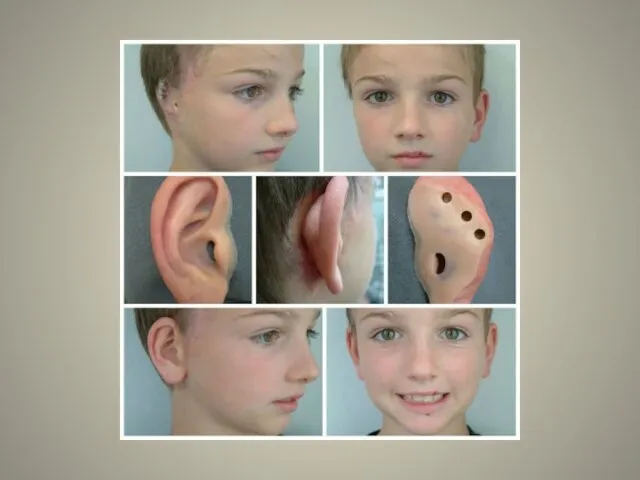
аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
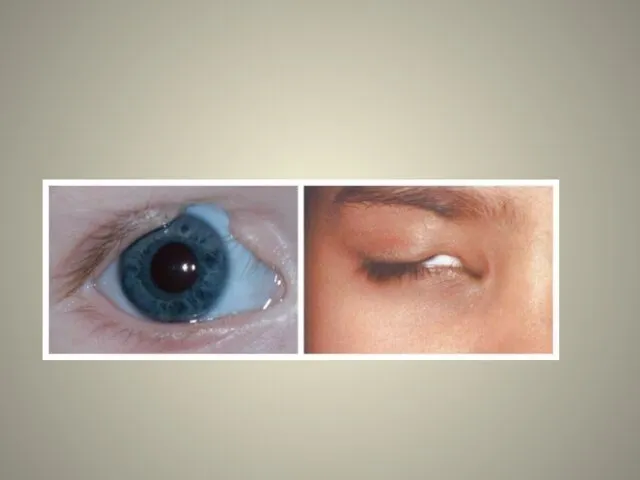
аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии.
Слайд 10Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации

OMENS, в которой выделяют пять групп аномалий:
O — поражение глазницы;
M — недоразвитие нижней челюсти;
E — аномалия уха;
N — вовлечённость нерва;
S — дефицит мягких тканей.
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован
Слайд 11Осложнения.
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей

деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне.
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [
Слайд 12Диагностика
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза

основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
КТ височных костей. На пораженной стороне выявляются грубые аномалии развития височной кости – уменьшение в размерах барабанной полости, конгломераты слуховых косточек, стеноз ниши окна преддверия, дистопия канала лицевого нерва.
Обследование у специалистов – генетика, кардиолога, ортопеда, офтальмолога, невропатолога, дефектолога, челюстно-лицевого хирурга, логопеда.
Дифференциальная диагностика с челюстно-лицевым дизостозом (синдромом Тричера Коллинза)
Слайд 13 Обязательной частью УЗИ плода на 18-20 неделе беременности является оценка лицевых структур. Методика

исследования включает мультиплоскостную оценку положения и размеров орбит, структур глазных яблок, правильности формирования челюстей, носа и носогубного треугольника, расположения и формы ушных раковин. УЗИ лицевых структур плода позволяет выявить недоразвитие нижней челюсти, расщелины губы и/или нёба, отсутствие или недоразвитие глаза и ушной раковины
Слайд 14Лечение
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся

в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
Слайд 16Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить

отставание ребёнка в речевом и общем развитии.
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами
Слайд 18Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений,

времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 3%.
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки
Слайд 19Список литературы
Беляков Ю. А. Стоматологические проявления наследственных болезней и синдромов. — М.:

Медицина, 1993.
Козлова С. И., Семанова Е., Демикова Н. С., Блинникова О. Е. Наследственные синдромы и медико-генетическое консультирование: справочник. — Л.: Медицина, 1989.
Андреева О. В., Анохина А. В., Краснов М. В., Загребаева Е. А. и др. Медико-генетическое консультирование в стоматологии // Вестник Чувашского университета. — 2011. — № 3. — С. 262-268.



 Спазмофилия
Спазмофилия The history of surgery
The history of surgery Amnesia. Types of amnesia
Amnesia. Types of amnesia Сосудистая эпилепсия
Сосудистая эпилепсия Методы исследования в гигиенической практике. Санитария. Структура Роспотребнадзора
Методы исследования в гигиенической практике. Санитария. Структура Роспотребнадзора Легенькая
Легенькая Личность безопасного типа поведения
Личность безопасного типа поведения Адам эмбриологиясы, жыныс жасушалары, ұрықтану, бөлшектену
Адам эмбриологиясы, жыныс жасушалары, ұрықтану, бөлшектену Народная медицина
Народная медицина Артикуляция и окклюзия. Виды окклюзии
Артикуляция и окклюзия. Виды окклюзии Виды наркоза. Основные препараты. Занятие 4
Виды наркоза. Основные препараты. Занятие 4 Комбинированные и хронические лучевые поражения
Комбинированные и хронические лучевые поражения Венеральды аурулар
Венеральды аурулар Методы обследования внутриутробного состояния плода
Методы обследования внутриутробного состояния плода Мочевыделительная система
Мочевыделительная система Воздушно-капельные инфекции
Воздушно-капельные инфекции Дифузний токс. зоб ІІ ст. сер.(пальпується, але не візуалізується при норм.положенні голови
Дифузний токс. зоб ІІ ст. сер.(пальпується, але не візуалізується при норм.положенні голови Матрица оценки эпидемиологической ситуации в регионах Казахстана
Матрица оценки эпидемиологической ситуации в регионах Казахстана Вазоспастическая стенокардия
Вазоспастическая стенокардия Хронический панкреатит
Хронический панкреатит Понятие травмы. Виды травм
Понятие травмы. Виды травм Методические основы патологической анатомии
Методические основы патологической анатомии Прокоагулянты. Классификация. Механизм действия. Показания к применению
Прокоагулянты. Классификация. Механизм действия. Показания к применению Инфекционные болезни
Инфекционные болезни Тамыр ішіне тамшылап құюдың техникасы
Тамыр ішіне тамшылап құюдың техникасы Инциденталомы надпочечников
Инциденталомы надпочечников Клинический случай
Клинический случай Особенности сестринского ухода при аллергических реакциях немедленного типа
Особенности сестринского ухода при аллергических реакциях немедленного типа