- Химическая термодинамика

Содержание
- 2. При изучении любых химических систем используют термодинамический и кинетический подход. В термодинамическом подходе рассматривают конечное и
- 3. Термодинамическая система – тело или группа тел, которые нас интересуют, все остальное – окружающая среда. По
- 4. Первое Начало утверждает, что теплота, переданная системе, идет на увеличение ее внутренней энергии и на работу
- 5. ИЗМЕНЕНИЕ ЭНТАЛЬПИИ СИСТЕМЫ В ХОДЕ ХИМИЧЕСКИХ РЕАКЦИЙ
- 6. Закон Кирхгофа гласит, что температурный коэффициент теплового эффекта химической реакции равен изменению теплоемкости системы в ходе
- 7. Поскольку многие химические процессы являются обратимыми, т.е. протекают в прямом и обратном направлении, необходимо помнить, что
- 8. НАПРАВЛЕННОСТЬ САМОПРОИЗВОЛЬНОГО ПРОТЕКАНИЯ ПРОЦЕССА Стремление системы к минимуму энергии – не единственная причина возможности самопроизвольного протекания
- 9. S = k ln W S – энтропия (функция состояния), от греч.trope – обращение, изменение. Размерность
- 10. ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ. ЭНЕРГИЯ ГИББСА . В ИЗОЛИРОВАННОЙ СИСТЕМЕ ВОЗМОЖНЫ ТОЛЬКО ТАКИЕ ПРОЦЕССЫ, КОТОРЫЕ СОПРОВОЖДАЮТСЯ РОСТОМ
- 11. ЭНЕРГИЯ ГИББСА
- 13. Скачать презентацию
Слайд 2При изучении любых химических систем используют термодинамический и кинетический подход. В термодинамическом
При изучении любых химических систем используют термодинамический и кинетический подход. В термодинамическом

Слайд 3Термодинамическая система – тело или группа тел, которые нас интересуют, все остальное
Термодинамическая система – тело или группа тел, которые нас интересуют, все остальное

Слайд 4Первое Начало утверждает, что теплота, переданная системе, идет на увеличение ее внутренней
Первое Начало утверждает, что теплота, переданная системе, идет на увеличение ее внутренней

Слайд 5ИЗМЕНЕНИЕ ЭНТАЛЬПИИ СИСТЕМЫ В ХОДЕ ХИМИЧЕСКИХ РЕАКЦИЙ
ИЗМЕНЕНИЕ ЭНТАЛЬПИИ СИСТЕМЫ В ХОДЕ ХИМИЧЕСКИХ РЕАКЦИЙ
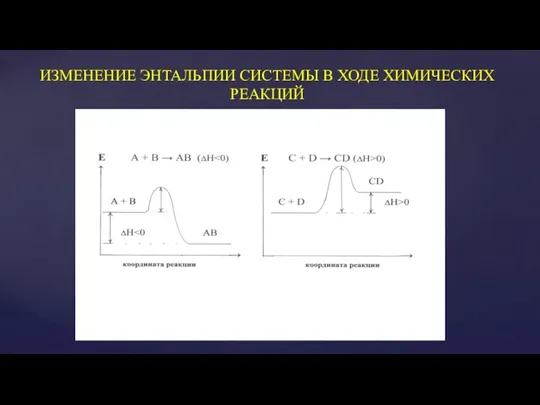
Слайд 6Закон Кирхгофа гласит, что температурный коэффициент теплового эффекта химической реакции равен изменению
Закон Кирхгофа гласит, что температурный коэффициент теплового эффекта химической реакции равен изменению

Слайд 7Поскольку многие химические процессы являются обратимыми, т.е. протекают в прямом и обратном
Поскольку многие химические процессы являются обратимыми, т.е. протекают в прямом и обратном

Слайд 8 НАПРАВЛЕННОСТЬ САМОПРОИЗВОЛЬНОГО ПРОТЕКАНИЯ ПРОЦЕССА
Стремление системы к минимуму энергии – не единственная
НАПРАВЛЕННОСТЬ САМОПРОИЗВОЛЬНОГО ПРОТЕКАНИЯ ПРОЦЕССА Стремление системы к минимуму энергии – не единственная

Слайд 9S = k ln W
S – энтропия (функция состояния), от греч.trope
S = k ln W
S – энтропия (функция состояния), от греч.trope

Слайд 10ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ. ЭНЕРГИЯ ГИББСА
.
В ИЗОЛИРОВАННОЙ СИСТЕМЕ ВОЗМОЖНЫ ТОЛЬКО ТАКИЕ ПРОЦЕССЫ, КОТОРЫЕ
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ. ЭНЕРГИЯ ГИББСА
.
В ИЗОЛИРОВАННОЙ СИСТЕМЕ ВОЗМОЖНЫ ТОЛЬКО ТАКИЕ ПРОЦЕССЫ, КОТОРЫЕ

Слайд 11
ЭНЕРГИЯ ГИББСА
ЭНЕРГИЯ ГИББСА

 Строение и функции нуклеиновых кислот. Лекция №3
Строение и функции нуклеиновых кислот. Лекция №3 Применение арт-технологии на уроках химии Обобщение опыта педагогической деятельности МОУ СОШ № 14 город Мончегорск Мурманск
Применение арт-технологии на уроках химии Обобщение опыта педагогической деятельности МОУ СОШ № 14 город Мончегорск Мурманск Буферные системы
Буферные системы Применение каучука
Применение каучука Классификация органических веществ
Классификация органических веществ Алкены
Алкены Соли, их классификация и свойства
Соли, их классификация и свойства Оксиды в свете ТЭД
Оксиды в свете ТЭД Кремний. Технический кремний
Кремний. Технический кремний Химия неорганическая и органическая
Химия неорганическая и органическая Презентация на тему Альдегиды и Кетоны
Презентация на тему Альдегиды и Кетоны  кристал решетка
кристал решетка Удивительная соль
Удивительная соль Периодическая система элементов, строение атома
Периодическая система элементов, строение атома Силикатная промышленность
Силикатная промышленность Эвапориты. Химические осадки из ионных растворов
Эвапориты. Химические осадки из ионных растворов Рівноваги у розчинах електролітів
Рівноваги у розчинах електролітів Натрий, свойства атома, химические и физические свойства
Натрий, свойства атома, химические и физические свойства Презентация на тему Альдегиды, свойства, получение, применение
Презентация на тему Альдегиды, свойства, получение, применение  Основные классы неорганических соединений. Лекция №3
Основные классы неорганических соединений. Лекция №3 Выращивание кристаллов соли и сахара в домашних условиях
Выращивание кристаллов соли и сахара в домашних условиях Презентация на тему Сплавы металлов
Презентация на тему Сплавы металлов  Азотная кислота и ее соли. Специфические свойства азотной кислоты
Азотная кислота и ее соли. Специфические свойства азотной кислоты Сигнальные молекулы
Сигнальные молекулы 4 Минеральные удобрения (1)
4 Минеральные удобрения (1) Ресурс Лови оксиды для систематизации знаний, умений, навыков по теме Оксиды
Ресурс Лови оксиды для систематизации знаний, умений, навыков по теме Оксиды Презентация на тему Бензол
Презентация на тему Бензол  Проверочные работы по темам: Неметаллы. 9 класс
Проверочные работы по темам: Неметаллы. 9 класс