Инфекционные процессы головного мозга в основе резистентных эпилепсий – современные подходы к диагностике и лечению
- Инфекционные процессы головного мозга в основе резистентных эпилепсий – современные подходы к диагностике и лечению

Содержание
- 2. Одной из причин симптоматических эпилепсий, резистентных к проводимой антиэпилептической терапии являются недиагностированные хронические энцефалиты и медленные
- 3. Энцефалит - заболевание головного мозга воспалительного характера с развитием инфекционного или инфекционно-аллергического процесса, обусловленного вирусами (чаще),
- 4. Классификация В связи с сложностью классификации энцефалитов по этиологии принято делить энцефалиты по механизму повреждающего действия
- 5. Первичные энцефалиты - механизм воздействия- непосредственное повреждение вирусом нейронов, эндотелия сосудов и оболочек мозга, приводящее к
- 6. К вопросам классификации энцефалитов НИ, которые могут принимать хроническое течение: клещевой (весенне-летний) энцефалит, эпидемический (Экономо), японский
- 7. Острые инфекции имеют обычно бурное начало, возникающую на 2-3 день выраженную неврологическую симптоматику, положительную динамику через
- 8. Особенности течения НИ Особенности течения инфекций в ЦНС определяются иммунологической "привилегированностью", анатомической обособленностью мозга, в частности:
- 9. На сегодняшний день установлено, что элементы иммунного ответа, связанные с течением аллергии, вносят важный вклад в
- 10. Иммунологические методы исследования при энцефалитах Уровень иммуноглобулинов G, A, M, E ЦИК (циркулирующие иммунные комплексы) Иммунофенотипирование
- 11. Клещевой энцефалит – хронизация Ежегодно в России регистрируется более 1000 случаев клещевого энцефалита. Хронический период КЭ
- 12. Эпилепсия Кожевникова Эпилепсия Кожевникова (ЭК) представляет полиэтиологичное заболевание, проявляющееся симптомокомплексом: облигатным является постоянный миоклонус, обычно сочетающийся
- 13. Эпилепсия Кожевникова «…сочетание генерализованных эпилептических приступов с постоянными клоническими судорогами в строго определенных частях тела. Из
- 14. Этиология ЭК (Walker & Shorvon в 1996 г.) Церебральные неоплазмы: метастазы, астроцитома, олигодендроглиома, карциноматоз Кортикальные дисплазии
- 15. Клещевой энцефалит В последние годы течение клещевого энцефалита претерпело изменения: Относительно редко стали наблюдаться тяжелые клинические
- 16. Диагностика в остром периоде КЭ Данные анамнеза Клиническая картина МРТ: диффузные корково-подкорковые атрофические изменения со вторичной
- 17. Лечение и профилактика КЭ Для специфического лечения применяют противоэнцефалитный донорский иммуноглобулин (в/м по 3—6 мл 1
- 18. Терапия Кожевниковской эпилепсии Антэпилептическая терапия: вальпроат 1000-2500 мг/сутки (30-80мг/кг) топирамат 75-400 мг/сутки (4-10 мг/кг/сутки) леветирацетам 1000-4000
- 19. Лирика (прегабалин): применение в клинической практике Показания В качестве вспомогательного средства у взрослых с парциальными судорогами,
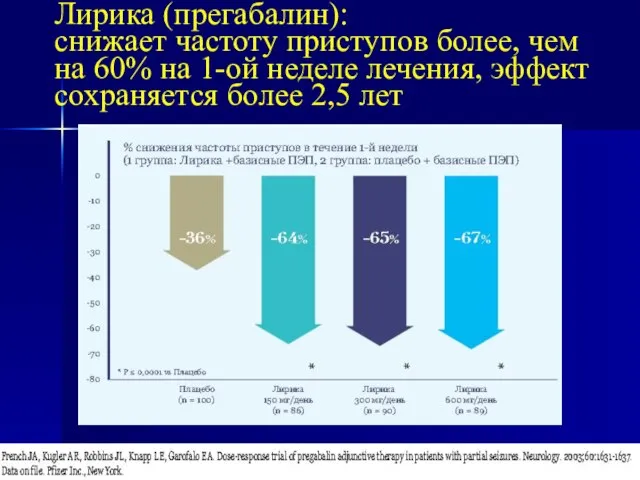
- 20. Лирика (прегабалин): снижает частоту приступов более, чем на 60% на 1-ой неделе лечения, эффект сохраняется более
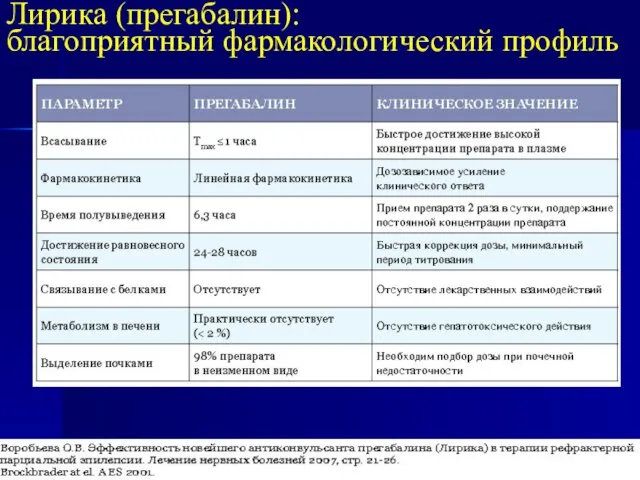
- 21. Лирика (прегабалин): благоприятный фармакологический профиль
- 22. Терапия Кожевниковской эпилепсии Лечение основного заболевания: хирургическое лечение сосудистая терапия нейропротекторная терапия иммуномодуляторы иммуносупрессоры стероидные гормоны
- 23. Нейропротекторные механизмы у различных препаратов Изменение метаболизма ряда нейромедиаторов и их способности к взаимодействию с мембранными
- 24. Нарушения высших психических функций в исходе энцефалита, вирус которого передаётся клещём Ещё в 1957 г. Feemster
- 25. Основное направление лечения - компенсаторная терапия - коррекция холинергического дефицита. Другое направление - применение антагонистов NMDA–рецепторов
- 26. Опыт кафедры Обследовано 7 пациентов с ЭК на фоне весенне-летнего КЭ (Кваскова Н.Е. с соавт., 2006)
- 27. Больной Е.Р., 7 лет. Диагноз: Хронический клещевой энцефалит. Симтоматическая фокальная эпилепсия. Кожевниковская форма. Правосторонний гемипарез Укус
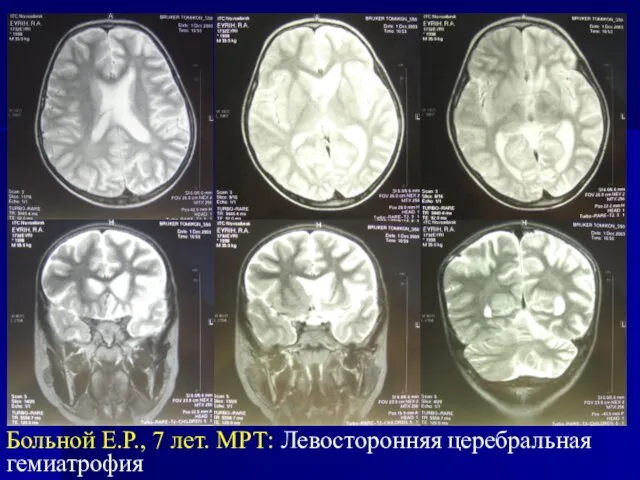
- 28. Больной Е.Р., 7 лет. МРТ: Левосторонняя церебральная гемиатрофия
- 29. Пациент Е.Р., 7 лет. ЭЭГ пробуждения: Разряды комплексов острая-медленная волна из левой лобной области, локальные с
- 30. Пациент Е.Р., 7 лет. ЭЭГ сна: Разряды в левой лобной области со склонностью к диффузному распространению
- 31. Энцефалит Расмуссена Хронический прогрессирующий очаговый энцефалит - хроническое заболевание головного мозга, проявляющееся фокальными моторными, миоклоническими, вторично-генерализованными
- 32. Энцефалит Расмуссена J.Bancaud и соавт. в 1992 году выделили три стадии клинического течения энцефалита Расмуссена: I
- 33. Диагностические критерии энцефалита Расмуссена (C.G.Bien, 2005) А: 1.Фокальные приступы (с/без эпилепсии Кожевникова) + односторонний кортикальный дефицит
- 34. Энцефалит Расмуссена ЭЭГ – в развёрнутой стадии выявляет нарушения в 100% случаев Наблюдается прогрессирующее замедление основной
- 35. Энцефалит Расмуссена Патологический процесс может распространяться на ствол мозга, полушария мозжечка, а также захватывать вторую гемисферу
- 36. Энцефалит Расмуссена В ряде случаев заболевание может возникать на фоне нейровисцеральной формы острой перемежающейся порфирии, которая
- 37. Отличия эпилепсии Кожевникова при клещевом энцефалите и энцефалите Расмуссена
- 38. Энцефалит Расмуссена Терапия: по мнению большинства авторов, наиболее эффективным методом лечения энцефалита Расмуссена является нейрохирургическое вмешательство
- 39. Больной П.Г., 16 лет. Диагноз: Энцефалит Расмуссена (?). Симптоматическая лобная эпилепсия. Жалобы: на приступы миоклонических подёргиваний
- 40. Клинический пример
- 41. Болезнь Лайма ЛБ (Лайм-боррелиоз, иксодовый боррелиоз) - распространенное природно-очаговое, бактериальное заболевание с трансмиссивным путем передачи, нередко
- 42. Диагностика в остром периоде БЛ Данные анамнеза Клиническая картина МРТ/КТ: диффузные корково-подкорковые атрофические изменения со вторичной
- 43. Неврологические, включая хронические проявления, болезни Лайма энцефалопатия с рассеянной органической симптоматикой, интеллектуально-мнестическими дефицитом менингорадикулит энцефаломиелит паркинсонический
- 44. Латентные проявления нейроборрелиоза Существуют латентные, клинически бессимптомные формы заболевания: Больные с повышенными сывороточными титрами IgG к
- 45. Терапия хронических проявлений нейроборрелиоза Симптоматическая терапия неврологических нарушений (энцефалопатия с рассеянной органической симптоматикой, поражения периферической нервной
- 46. Терапия симптоматической эпилепсии при БЛ карбамазепин 15-35 мг/кг/сутки окскарбазепин (стартовая доза с 5 мг/кг/сутки, максимальная доза
- 47. Энцефалит Экономо В хронической стадии наряду с синдромом паркинсонизма и эндокринными расстройствами (адипозогенитальная дистрофия, инфантилизм, нарушение
- 48. Терапия хронических проявлений энцефалита Экономо Симптоматическая терапия неврологических нарушений (нейроэндокринных расстройств, когнитивных расстройств, паркинсонического синдрома, психоорганического
- 49. Энцефалит Давсона (ПСПЭ) Энцефалит с включениями Давсона, узелковый панэнцефалит Петте - Деринга, лейкоэнцефалит Ван-Богарта) Впервые упоминается
- 50. ПСПЭ ПСПЭ - очень медленнотекущая форма коревого энцефалита Чаще всего возникает у лиц, переболевших в детстве
- 51. ПСПЭ сложно диагностировать на начальных стадиях заболевания, вероятность ошибочного диагноза высока (G. Gallucci, 2001). Ранее это
- 52. ПСПЭ Антитела к вирусу кори представлены классами иммуноглобулинов IgG, IgM, IgA, наибольшая часть антител в СМЖ
- 53. Клиническая картина ПСП Стадии ПСПЭ на основе обобщения классификаций, предложенных J. Jabbour и W. Risk и
- 54. Стадия 2. Дебютируют судороги: фокальные, генерализованные тонико-клонические, миоклонические Характерно расстройство праксиса Зрительные нарушения у 50% пациентов
- 55. Диагностика ПСПЭ Клиническая картина (4 стадии) ЭЭГ: характерны периодические разряды (каждые 3-8 сек) высокоамплитудных остроконечных медленных
- 56. Клинический пример Больной П.Д., 14 лет Диагноз: Подострый склерозирующий панэнцефалит Ван-Богарта. Правосторонний гемипарез, подкорковый синдром, псевдобульбарный
- 57. Клинический пример Из анамнеза жизни: ребенок от 1 беременности, протекавшей физиологично, роды срочные, раннее развитие по
- 58. Клинический пример ЭЭГ: На всем протяжении записи отмечались кластерные иктальные паттерны - диффузных синхронизированных всплесков медленных
- 59. Клинический пример МРТ головного мозга больного П.Д. Обширная зона поражения преимущественно белого вещества, больше слева, в
- 60. Клинический пример
- 61. Клинический пример Консультация генетика в МГНЦ (д.м.н., профессор Дадали Е.Л.): данных за наследственную патологию недостаточно. В
- 62. Терапия ПСП Противовирусные препараты: изопринозин (инозиплекс), рибавирин, амантадин, циметидин Иммуномодуляторы: интерферон альфа-2а (роферон), в том числе
- 63. Прогрессирующий краснушный панэнцефалит Крайне редкое заболевание - в основном является следствием врожденной краснухи, но может возникнуть
- 64. Прогрессирующий краснушный панэнцефалит ЦСЖ: легкий лимфоцитоз (менее 40 в мкл), небольшое повышение концентрации белка (менее 1,5
- 65. Лимбический аутоиммунный энцефалит Поражает преимущественно структуры лимбической системы, сопровождается системным нарушением вегетативных функций. Выраженный мнестический дефицит
- 66. Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит) Тяжелая эпилептическая энцефалопатия школьного возраста (DEVASTATING EPILEPTIC ENCEPHALOPATHY IN SCHOOL-AGED
- 67. Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит) Диагностические критерии: (Y. Mikaeloff et al., 2008): дебют в 4-11
- 68. Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит) Терапия: в остром периоде – купирование эпистатуса по протоколу (крайняя
- 69. Трансмиссивные спонгиформные энцефалопатии (ТСЭ) – это уникальные прионовые заболевания животных и людей, которые могут быть наследственными,
- 70. Спонгиформные трансмиссивные энцефалопатии куру – у людей бычья спонгиоформная энцефалопатия (БСЭ) – у крупного рогатого скота
- 71. Губкообразная энцефалопатия крупного рогатого скота Коровье бешенство — нейродегенеративная прионная болезнь, приводящая к необратимым, летальным изменениям
- 72. Фатальная семейная бессонница Fatal family insomnia, FFI — редкое неизлечимое наследственное заболевание, при котором больной умирает
- 73. Фатальная семейная бессонница Клинически выделяют 4 стадии заболевания: Первые 4 месяца характеризуются нарастающей бессонницей с паническими
- 74. С-м Герстманна-Штраусслера-Шейнкера Gerstmann-Sträussler-Scheinker syndrome (подострая губчатая энцефалопатия, тип Герштмана-Штрауслера (subacute spongiform encephalopaty, Gerstmann-Sträussler type) - относится
- 75. С-м Герстманна-Штраусслера-Шейнкера Считается, что заболевание встречается у 1-10 человек на каждые 100 миллионов. Встречаемость 1 случай
- 76. Болезнь Крейтцфельдта-Якоба Прионы распространяются в нервной системе, тканях глазаПрионы распространяются в нервной системе, тканях глаза и
- 77. Наиболее известной из прион-ассоциированных заболеваний является болезнь Крейцфельда-Якоба, которая представлена следующими формами: спорадическая семейная ятрогенная новый
- 78. 1. Продромальный период — симптомы неспецифичны и возникают примерно у 30% больных. Они появляются за недели
- 79. Диагностика БКЯ Определенная БКЯ: • характерная неврологическая и морфологическая в том числе патолого-анатомическая и нейрорадиологическая симптоматика
- 80. Диагностика БКЯ характерные данные магнитно-резонансной томографии (МРТ) головного мозга (особенно на поздних стадиях развития заболевания) позитронно-эмиссионая
- 81. Диагностика БКЯ
- 82. Диагностика БКЯ Анализ СМЖ (люмбальная пункция должна проводиться во всех случаях БКЯ) – м.б. небольшое повышение
- 83. Лечение прионовых заболеваний необходимо отменить все лекарственные препараты, которые могут негативно влиять на мнестические функции и
- 84. Лечение БКЯ и других прионовых заболеваний БКЯ характеризуется отсутствием иммунного ответа на прионовую инфекцию. Это связано
- 85. Резистентные эпилепсии Симптоматическая фокальная эпилепсия составляет 52% от всех форм эпилепсии. При фокальной эпилепсии доля резистентных
- 86. Критерии резистентности Отсутствие эффективности при терапии базовыми АЭП в возрастных дозировках, снижение числа приступов менее чем
- 87. Морфофункциональная основа резистентной эпилепсии Участки фокальных корковых дисплазий генерируют высокоамплитудные аномальной модальности биопотенциалы, которые пробивают нейронные
- 88. Причины резистентности Объективные Грубый структурный дефект головного мозга Прогрессирование неврологического заболевания (дегенерации, хронические энцефалиты, медленные НИ)
- 90. Скачать презентацию
Слайд 2Одной из причин симптоматических эпилепсий, резистентных к проводимой антиэпилептической терапии являются недиагностированные
Одной из причин симптоматических эпилепсий, резистентных к проводимой антиэпилептической терапии являются недиагностированные

Слайд 3Энцефалит - заболевание головного мозга воспалительного характера с развитием инфекционного или инфекционно-аллергического
Энцефалит - заболевание головного мозга воспалительного характера с развитием инфекционного или инфекционно-аллергического

Слайд 4Классификация
В связи с сложностью классификации энцефалитов по этиологии принято делить энцефалиты по
Классификация
В связи с сложностью классификации энцефалитов по этиологии принято делить энцефалиты по

Слайд 5Первичные энцефалиты - механизм воздействия- непосредственное повреждение вирусом нейронов, эндотелия сосудов и
Первичные энцефалиты - механизм воздействия- непосредственное повреждение вирусом нейронов, эндотелия сосудов и
Слайд 6К вопросам классификации энцефалитов
НИ, которые могут принимать хроническое течение: клещевой (весенне-летний) энцефалит,
К вопросам классификации энцефалитов
НИ, которые могут принимать хроническое течение: клещевой (весенне-летний) энцефалит,

Слайд 7Острые инфекции имеют обычно бурное начало, возникающую на 2-3 день выраженную неврологическую

Слайд 8Особенности течения НИ
Особенности течения инфекций в ЦНС определяются иммунологической "привилегированностью", анатомической обособленностью
Особенности течения НИ
Особенности течения инфекций в ЦНС определяются иммунологической "привилегированностью", анатомической обособленностью

Слайд 9На сегодняшний день установлено, что элементы иммунного ответа, связанные с течением аллергии,
На сегодняшний день установлено, что элементы иммунного ответа, связанные с течением аллергии,

Слайд 10Иммунологические методы
исследования при энцефалитах
Уровень иммуноглобулинов G, A, M, E
ЦИК
Иммунологические методы
исследования при энцефалитах
Уровень иммуноглобулинов G, A, M, E
ЦИК

Слайд 11Клещевой энцефалит – хронизация
Ежегодно в России регистрируется более 1000 случаев клещевого энцефалита.
Хронический
Клещевой энцефалит – хронизация
Ежегодно в России регистрируется более 1000 случаев клещевого энцефалита.
Хронический

Слайд 12Эпилепсия Кожевникова
Эпилепсия Кожевникова (ЭК) представляет полиэтиологичное заболевание, проявляющееся симптомокомплексом:
облигатным является
Эпилепсия Кожевникова
Эпилепсия Кожевникова (ЭК) представляет полиэтиологичное заболевание, проявляющееся симптомокомплексом:
облигатным является

Слайд 13Эпилепсия Кожевникова
«…сочетание генерализованных эпилептических приступов с постоянными клоническими судорогами в строго определенных
Эпилепсия Кожевникова
«…сочетание генерализованных эпилептических приступов с постоянными клоническими судорогами в строго определенных

Слайд 14Этиология ЭК (Walker & Shorvon в 1996 г.)
Церебральные неоплазмы: метастазы, астроцитома, олигодендроглиома,
Этиология ЭК (Walker & Shorvon в 1996 г.)
Церебральные неоплазмы: метастазы, астроцитома, олигодендроглиома,

Слайд 15Клещевой энцефалит
В последние годы течение клещевого энцефалита претерпело изменения:
Относительно редко стали
Клещевой энцефалит
В последние годы течение клещевого энцефалита претерпело изменения:
Относительно редко стали

Слайд 16Диагностика в остром периоде КЭ
Данные анамнеза
Клиническая картина
МРТ: диффузные корково-подкорковые атрофические изменения
Диагностика в остром периоде КЭ
Данные анамнеза
Клиническая картина
МРТ: диффузные корково-подкорковые атрофические изменения

Слайд 17Лечение и профилактика КЭ
Для специфического лечения применяют противоэнцефалитный донорский иммуноглобулин (в/м
Лечение и профилактика КЭ
Для специфического лечения применяют противоэнцефалитный донорский иммуноглобулин (в/м

Слайд 18Терапия Кожевниковской эпилепсии
Антэпилептическая терапия:
вальпроат 1000-2500 мг/сутки (30-80мг/кг)
топирамат 75-400 мг/сутки (4-10 мг/кг/сутки)
Терапия Кожевниковской эпилепсии
Антэпилептическая терапия:
вальпроат 1000-2500 мг/сутки (30-80мг/кг)
топирамат 75-400 мг/сутки (4-10 мг/кг/сутки)

Слайд 19Лирика (прегабалин): применение в клинической практике
Показания
В качестве вспомогательного средства у взрослых с
Лирика (прегабалин): применение в клинической практике
Показания
В качестве вспомогательного средства у взрослых с

Слайд 20Лирика (прегабалин):
снижает частоту приступов более, чем на 60% на 1-ой неделе лечения,
Лирика (прегабалин): снижает частоту приступов более, чем на 60% на 1-ой неделе лечения,
Слайд 21Лирика (прегабалин):
благоприятный фармакологический профиль
Лирика (прегабалин):
благоприятный фармакологический профиль
Слайд 22Терапия Кожевниковской эпилепсии
Лечение основного заболевания:
хирургическое лечение
сосудистая терапия
нейропротекторная терапия
иммуномодуляторы
иммуносупрессоры
стероидные гормоны
Терапия Кожевниковской эпилепсии
Лечение основного заболевания:
хирургическое лечение
сосудистая терапия
нейропротекторная терапия
иммуномодуляторы
иммуносупрессоры
стероидные гормоны

Слайд 23Нейропротекторные механизмы у
различных препаратов
Изменение метаболизма ряда нейромедиаторов и их способности к
Нейропротекторные механизмы у
различных препаратов
Изменение метаболизма ряда нейромедиаторов и их способности к

Слайд 24Нарушения высших психических функций в исходе энцефалита, вирус которого передаётся клещём
Ещё в
Нарушения высших психических функций в исходе энцефалита, вирус которого передаётся клещём
Ещё в

Слайд 25Основное направление лечения - компенсаторная терапия - коррекция холинергического дефицита.
Другое направление
Основное направление лечения - компенсаторная терапия - коррекция холинергического дефицита.
Другое направление

Слайд 26Опыт кафедры
Обследовано 7 пациентов с ЭК на фоне весенне-летнего КЭ (Кваскова Н.Е.
Опыт кафедры
Обследовано 7 пациентов с ЭК на фоне весенне-летнего КЭ (Кваскова Н.Е.

Слайд 27Больной Е.Р., 7 лет. Диагноз: Хронический клещевой энцефалит. Симтоматическая фокальная эпилепсия. Кожевниковская
Больной Е.Р., 7 лет. Диагноз: Хронический клещевой энцефалит. Симтоматическая фокальная эпилепсия. Кожевниковская

Слайд 28Больной Е.Р., 7 лет. МРТ: Левосторонняя церебральная гемиатрофия
Больной Е.Р., 7 лет. МРТ: Левосторонняя церебральная гемиатрофия
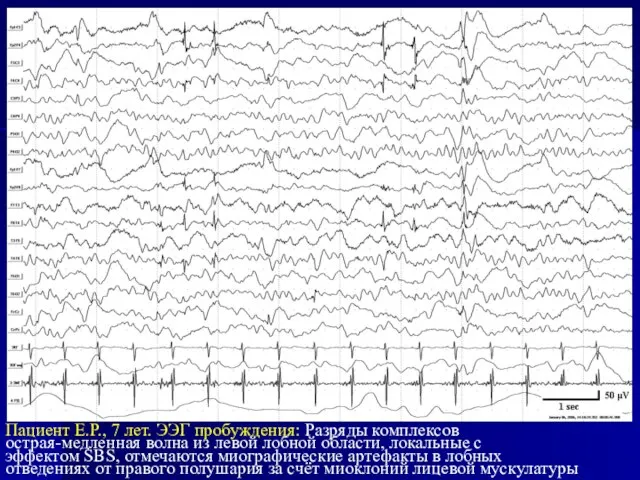
Слайд 29Пациент Е.Р., 7 лет. ЭЭГ пробуждения: Разряды комплексов
острая-медленная волна из левой
Пациент Е.Р., 7 лет. ЭЭГ пробуждения: Разряды комплексов
острая-медленная волна из левой
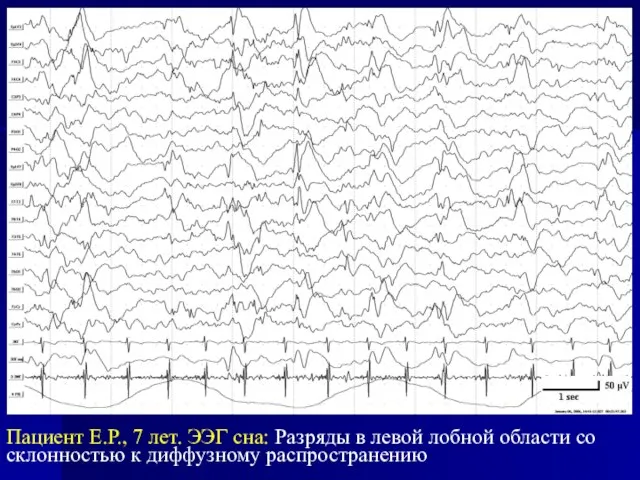
Слайд 30Пациент Е.Р., 7 лет. ЭЭГ сна: Разряды в левой лобной области со
Пациент Е.Р., 7 лет. ЭЭГ сна: Разряды в левой лобной области со
Слайд 31Энцефалит Расмуссена
Хронический прогрессирующий очаговый энцефалит - хроническое заболевание головного мозга, проявляющееся фокальными
Энцефалит Расмуссена
Хронический прогрессирующий очаговый энцефалит - хроническое заболевание головного мозга, проявляющееся фокальными

Слайд 32Энцефалит Расмуссена
J.Bancaud и соавт. в 1992 году выделили три стадии клинического течения
Энцефалит Расмуссена
J.Bancaud и соавт. в 1992 году выделили три стадии клинического течения

Слайд 33Диагностические критерии энцефалита Расмуссена (C.G.Bien, 2005)
А:
1.Фокальные приступы (с/без эпилепсии Кожевникова) + односторонний
Диагностические критерии энцефалита Расмуссена (C.G.Bien, 2005)
А:
1.Фокальные приступы (с/без эпилепсии Кожевникова) + односторонний

Слайд 34Энцефалит Расмуссена
ЭЭГ – в развёрнутой стадии выявляет нарушения в 100% случаев
Наблюдается прогрессирующее
Энцефалит Расмуссена
ЭЭГ – в развёрнутой стадии выявляет нарушения в 100% случаев
Наблюдается прогрессирующее

Слайд 35Энцефалит Расмуссена
Патологический процесс может распространяться на ствол мозга, полушария мозжечка, а также
Энцефалит Расмуссена
Патологический процесс может распространяться на ствол мозга, полушария мозжечка, а также

Слайд 36Энцефалит Расмуссена
В ряде случаев заболевание может возникать на фоне нейровисцеральной формы острой
Энцефалит Расмуссена
В ряде случаев заболевание может возникать на фоне нейровисцеральной формы острой

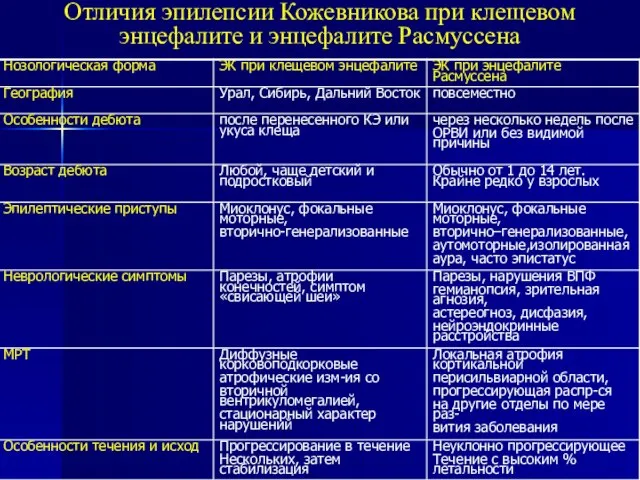
Слайд 37Отличия эпилепсии Кожевникова при клещевом энцефалите и энцефалите Расмуссена
Отличия эпилепсии Кожевникова при клещевом энцефалите и энцефалите Расмуссена
Слайд 38Энцефалит Расмуссена
Терапия: по мнению большинства авторов, наиболее эффективным методом лечения энцефалита Расмуссена
Энцефалит Расмуссена
Терапия: по мнению большинства авторов, наиболее эффективным методом лечения энцефалита Расмуссена

Слайд 39Больной П.Г., 16 лет. Диагноз: Энцефалит Расмуссена (?). Симптоматическая лобная эпилепсия.
Жалобы:
Больной П.Г., 16 лет. Диагноз: Энцефалит Расмуссена (?). Симптоматическая лобная эпилепсия. Жалобы:

Слайд 40Клинический пример
Клинический пример
Слайд 41Болезнь Лайма
ЛБ (Лайм-боррелиоз, иксодовый боррелиоз) - распространенное природно-очаговое, бактериальное заболевание с трансмиссивным
Болезнь Лайма
ЛБ (Лайм-боррелиоз, иксодовый боррелиоз) - распространенное природно-очаговое, бактериальное заболевание с трансмиссивным

Слайд 42Диагностика в остром периоде БЛ
Данные анамнеза
Клиническая картина
МРТ/КТ: диффузные корково-подкорковые атрофические изменения со
Диагностика в остром периоде БЛ
Данные анамнеза
Клиническая картина
МРТ/КТ: диффузные корково-подкорковые атрофические изменения со

Слайд 43Неврологические, включая хронические проявления, болезни Лайма
энцефалопатия с рассеянной органической симптоматикой, интеллектуально-мнестическими дефицитом
Неврологические, включая хронические проявления, болезни Лайма
энцефалопатия с рассеянной органической симптоматикой, интеллектуально-мнестическими дефицитом

Слайд 44Латентные проявления нейроборрелиоза
Существуют латентные, клинически бессимптомные формы заболевания:
Больные с повышенными сывороточными
Латентные проявления нейроборрелиоза
Существуют латентные, клинически бессимптомные формы заболевания:
Больные с повышенными сывороточными

Слайд 45Терапия хронических проявлений нейроборрелиоза
Симптоматическая терапия неврологических нарушений (энцефалопатия с рассеянной органической симптоматикой,
Терапия хронических проявлений нейроборрелиоза
Симптоматическая терапия неврологических нарушений (энцефалопатия с рассеянной органической симптоматикой,

Слайд 46Терапия симптоматической эпилепсии при БЛ
карбамазепин 15-35 мг/кг/сутки
окскарбазепин (стартовая доза с 5 мг/кг/сутки,
Терапия симптоматической эпилепсии при БЛ
карбамазепин 15-35 мг/кг/сутки
окскарбазепин (стартовая доза с 5 мг/кг/сутки,

Слайд 47Энцефалит Экономо
В хронической стадии наряду с синдромом паркинсонизма и эндокринными расстройствами (адипозогенитальная
Энцефалит Экономо
В хронической стадии наряду с синдромом паркинсонизма и эндокринными расстройствами (адипозогенитальная

Слайд 48Терапия хронических проявлений энцефалита Экономо
Симптоматическая терапия неврологических нарушений (нейроэндокринных расстройств, когнитивных расстройств,
Терапия хронических проявлений энцефалита Экономо
Симптоматическая терапия неврологических нарушений (нейроэндокринных расстройств, когнитивных расстройств,

Слайд 49Энцефалит Давсона (ПСПЭ)
Энцефалит с включениями Давсона, узелковый панэнцефалит Петте - Деринга, лейкоэнцефалит
Энцефалит Давсона (ПСПЭ)
Энцефалит с включениями Давсона, узелковый панэнцефалит Петте - Деринга, лейкоэнцефалит

Слайд 50ПСПЭ
ПСПЭ - очень медленнотекущая форма коревого энцефалита
Чаще всего возникает у лиц, переболевших
ПСПЭ
ПСПЭ - очень медленнотекущая форма коревого энцефалита
Чаще всего возникает у лиц, переболевших

Слайд 51ПСПЭ сложно диагностировать на начальных стадиях заболевания, вероятность ошибочного диагноза высока (G.
ПСПЭ сложно диагностировать на начальных стадиях заболевания, вероятность ошибочного диагноза высока (G.

Слайд 52ПСПЭ
Антитела к вирусу кори представлены классами иммуноглобулинов IgG, IgM, IgA, наибольшая часть
ПСПЭ
Антитела к вирусу кори представлены классами иммуноглобулинов IgG, IgM, IgA, наибольшая часть

Слайд 53Клиническая картина ПСП
Стадии ПСПЭ на основе обобщения классификаций, предложенных J. Jabbour и
Клиническая картина ПСП
Стадии ПСПЭ на основе обобщения классификаций, предложенных J. Jabbour и

Слайд 54Стадия 2.
Дебютируют судороги: фокальные, генерализованные тонико-клонические, миоклонические
Характерно расстройство праксиса
Зрительные нарушения
Стадия 2.
Дебютируют судороги: фокальные, генерализованные тонико-клонические, миоклонические
Характерно расстройство праксиса
Зрительные нарушения

Слайд 55Диагностика ПСПЭ
Клиническая картина (4 стадии)
ЭЭГ: характерны периодические разряды (каждые 3-8 сек) высокоамплитудных
Диагностика ПСПЭ
Клиническая картина (4 стадии)
ЭЭГ: характерны периодические разряды (каждые 3-8 сек) высокоамплитудных

Слайд 56Клинический пример
Больной П.Д., 14 лет
Диагноз: Подострый склерозирующий панэнцефалит Ван-Богарта. Правосторонний гемипарез, подкорковый
Клинический пример
Больной П.Д., 14 лет
Диагноз: Подострый склерозирующий панэнцефалит Ван-Богарта. Правосторонний гемипарез, подкорковый

Слайд 57Клинический пример
Из анамнеза жизни: ребенок от 1 беременности, протекавшей физиологично, роды срочные,
Клинический пример
Из анамнеза жизни: ребенок от 1 беременности, протекавшей физиологично, роды срочные,

Слайд 58Клинический пример
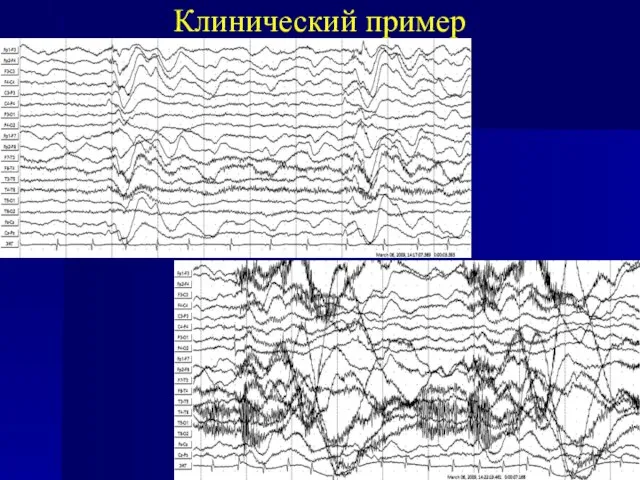
ЭЭГ: На всем протяжении записи отмечались кластерные иктальные паттерны - диффузных
Клинический пример
ЭЭГ: На всем протяжении записи отмечались кластерные иктальные паттерны - диффузных

Слайд 59Клинический пример
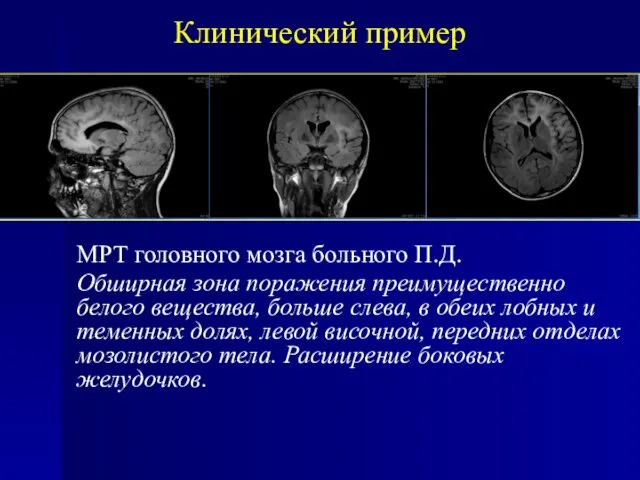
МРТ головного мозга больного П.Д.
Обширная зона поражения преимущественно белого вещества, больше
Клинический пример
МРТ головного мозга больного П.Д.
Обширная зона поражения преимущественно белого вещества, больше
Слайд 60Клинический пример
Клинический пример
Слайд 61Клинический пример
Консультация генетика в МГНЦ (д.м.н., профессор Дадали Е.Л.): данных за наследственную
Клинический пример
Консультация генетика в МГНЦ (д.м.н., профессор Дадали Е.Л.): данных за наследственную

Слайд 62Терапия ПСП
Противовирусные препараты: изопринозин (инозиплекс), рибавирин, амантадин, циметидин
Иммуномодуляторы: интерферон альфа-2а (роферон),
Терапия ПСП
Противовирусные препараты: изопринозин (инозиплекс), рибавирин, амантадин, циметидин
Иммуномодуляторы: интерферон альфа-2а (роферон),

Слайд 63Прогрессирующий краснушный панэнцефалит
Крайне редкое заболевание - в основном является следствием врожденной краснухи,
Прогрессирующий краснушный панэнцефалит
Крайне редкое заболевание - в основном является следствием врожденной краснухи,

Слайд 64Прогрессирующий краснушный панэнцефалит
ЦСЖ: легкий лимфоцитоз (менее 40 в мкл), небольшое повышение концентрации
Прогрессирующий краснушный панэнцефалит
ЦСЖ: легкий лимфоцитоз (менее 40 в мкл), небольшое повышение концентрации

Слайд 65Лимбический аутоиммунный энцефалит
Поражает преимущественно структуры
лимбической системы, сопровождается
системным нарушением вегетативных
функций. Выраженный
Лимбический аутоиммунный энцефалит
Поражает преимущественно структуры
лимбической системы, сопровождается
системным нарушением вегетативных
функций. Выраженный

Слайд 66Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит)
Тяжелая эпилептическая энцефалопатия школьного возраста
(DEVASTATING EPILEPTIC ENCEPHALOPATHY
Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит)
Тяжелая эпилептическая энцефалопатия школьного возраста
(DEVASTATING EPILEPTIC ENCEPHALOPATHY

Слайд 67Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит)
Диагностические критерии: (Y. Mikaeloff et al., 2008):
дебют
Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит)
Диагностические критерии: (Y. Mikaeloff et al., 2008):
дебют

Слайд 68Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит)
Терапия: в остром периоде – купирование эпистатуса
Тяжелая эпилептическая энцефалопатия школьного возраста (псевдоэнцефалит)
Терапия: в остром периоде – купирование эпистатуса

Слайд 69Трансмиссивные спонгиформные энцефалопатии (ТСЭ) – это уникальные прионовые заболевания животных и людей,
Трансмиссивные спонгиформные энцефалопатии (ТСЭ) – это уникальные прионовые заболевания животных и людей,

Слайд 70Спонгиформные трансмиссивные энцефалопатии
куру – у людей
бычья спонгиоформная энцефалопатия (БСЭ) – у крупного
Спонгиформные трансмиссивные энцефалопатии
куру – у людей
бычья спонгиоформная энцефалопатия (БСЭ) – у крупного

Слайд 71Губкообразная энцефалопатия крупного рогатого скота
Коровье бешенство — нейродегенеративная прионная болезнь, приводящая к
Губкообразная энцефалопатия крупного рогатого скота
Коровье бешенство — нейродегенеративная прионная болезнь, приводящая к

Слайд 72Фатальная семейная бессонница
Fatal family insomnia, FFI — редкое неизлечимое наследственное заболевание, при
Фатальная семейная бессонница
Fatal family insomnia, FFI — редкое неизлечимое наследственное заболевание, при

Слайд 73Фатальная семейная бессонница
Клинически выделяют 4 стадии заболевания:
Первые 4 месяца характеризуются нарастающей бессонницей
Фатальная семейная бессонница
Клинически выделяют 4 стадии заболевания:
Первые 4 месяца характеризуются нарастающей бессонницей

Слайд 74С-м Герстманна-Штраусслера-Шейнкера
Gerstmann-Sträussler-Scheinker syndrome (подострая губчатая энцефалопатия, тип Герштмана-Штрауслера (subacute spongiform encephalopaty, Gerstmann-Sträussler
С-м Герстманна-Штраусслера-Шейнкера
Gerstmann-Sträussler-Scheinker syndrome (подострая губчатая энцефалопатия, тип Герштмана-Штрауслера (subacute spongiform encephalopaty, Gerstmann-Sträussler

Слайд 75С-м Герстманна-Штраусслера-Шейнкера
Считается, что заболевание встречается у 1-10 человек на каждые 100 миллионов.
Встречаемость
С-м Герстманна-Штраусслера-Шейнкера
Считается, что заболевание встречается у 1-10 человек на каждые 100 миллионов.
Встречаемость

Слайд 76Болезнь Крейтцфельдта-Якоба
Прионы распространяются в нервной системе, тканях глазаПрионы распространяются в нервной системе,
Болезнь Крейтцфельдта-Якоба
Прионы распространяются в нервной системе, тканях глазаПрионы распространяются в нервной системе,

Слайд 77Наиболее известной из прион-ассоциированных заболеваний является болезнь Крейцфельда-Якоба, которая представлена следующими формами:
Наиболее известной из прион-ассоциированных заболеваний является болезнь Крейцфельда-Якоба, которая представлена следующими формами:
Слайд 781. Продромальный период — симптомы неспецифичны и возникают примерно у 30% больных.
1. Продромальный период — симптомы неспецифичны и возникают примерно у 30% больных.

Слайд 79Диагностика БКЯ
Определенная БКЯ:
• характерная неврологическая и морфологическая в том числе патолого-анатомическая и
Диагностика БКЯ
Определенная БКЯ: • характерная неврологическая и морфологическая в том числе патолого-анатомическая и

Слайд 80Диагностика БКЯ
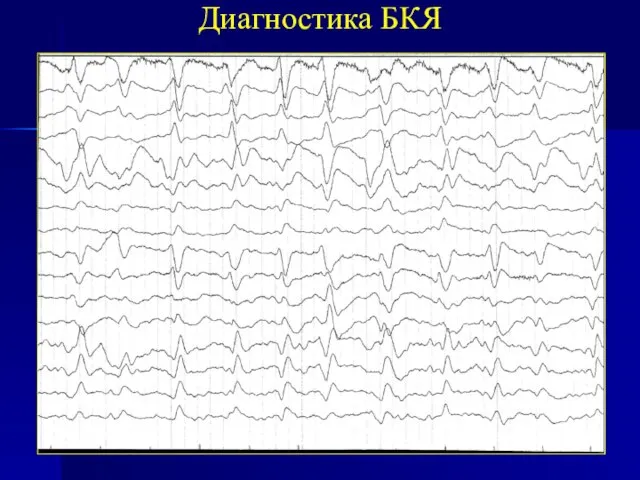
характерные данные магнитно-резонансной томографии (МРТ) головного мозга (особенно на поздних стадиях
Диагностика БКЯ
характерные данные магнитно-резонансной томографии (МРТ) головного мозга (особенно на поздних стадиях

Слайд 81Диагностика БКЯ
Диагностика БКЯ
Слайд 82Диагностика БКЯ
Анализ СМЖ (люмбальная пункция должна проводиться во всех случаях БКЯ) –
Диагностика БКЯ
Анализ СМЖ (люмбальная пункция должна проводиться во всех случаях БКЯ) –

Слайд 83Лечение прионовых заболеваний
необходимо отменить все лекарственные препараты, которые могут негативно влиять на
Лечение прионовых заболеваний
необходимо отменить все лекарственные препараты, которые могут негативно влиять на

Слайд 84Лечение БКЯ и других прионовых заболеваний
БКЯ характеризуется отсутствием иммунного ответа на прионовую
Лечение БКЯ и других прионовых заболеваний
БКЯ характеризуется отсутствием иммунного ответа на прионовую

Слайд 85Резистентные эпилепсии
Симптоматическая фокальная эпилепсия составляет 52% от всех форм эпилепсии. При фокальной
Резистентные эпилепсии
Симптоматическая фокальная эпилепсия составляет 52% от всех форм эпилепсии. При фокальной

Слайд 86Критерии резистентности
Отсутствие эффективности при терапии базовыми АЭП в возрастных дозировках, снижение числа
Критерии резистентности
Отсутствие эффективности при терапии базовыми АЭП в возрастных дозировках, снижение числа

Слайд 87Морфофункциональная основа резистентной эпилепсии
Участки фокальных корковых дисплазий генерируют высокоамплитудные аномальной модальности биопотенциалы,
Морфофункциональная основа резистентной эпилепсии
Участки фокальных корковых дисплазий генерируют высокоамплитудные аномальной модальности биопотенциалы,

Слайд 88Причины резистентности
Объективные
Грубый структурный дефект головного мозга
Прогрессирование неврологического заболевания (дегенерации, хронические энцефалиты,
Причины резистентности
Объективные
Грубый структурный дефект головного мозга
Прогрессирование неврологического заболевания (дегенерации, хронические энцефалиты,

 Shiatsu
Shiatsu Особенности физиологии новорожденных и слингоношение
Особенности физиологии новорожденных и слингоношение Основные хирургические навыки. Лекция 1
Основные хирургические навыки. Лекция 1 Поражение органов зрения и головного мозга у глубоко недоношенных детей
Поражение органов зрения и головного мозга у глубоко недоношенных детей Острый панкреатит. Анатомия поджелудочной железы
Острый панкреатит. Анатомия поджелудочной железы Стоит ли принимать ноотропы перед сессией. Группы ноотропов
Стоит ли принимать ноотропы перед сессией. Группы ноотропов Острая ревматическая лихорадка
Острая ревматическая лихорадка Расстройства пищевого поведения
Расстройства пищевого поведения Психолого-педагогическая характеристика глухих детей
Психолого-педагогическая характеристика глухих детей Оказание первой помощи у пациентов с ожогами кожных покровов
Оказание первой помощи у пациентов с ожогами кожных покровов Око як оптична система. Зір і бачення. Окуляри. Вади зору та їх корекція
Око як оптична система. Зір і бачення. Окуляри. Вади зору та їх корекція Инструментальные методы исследования
Инструментальные методы исследования Опухоли, бластомы, новообразования
Опухоли, бластомы, новообразования Выпускная квалификационная работа Особенности репродуктивного здоровья девушек-подростков, страдающих ожирением
Выпускная квалификационная работа Особенности репродуктивного здоровья девушек-подростков, страдающих ожирением Жаңашыл,креативті,педагог,шығармашыл,толерантты,медбике модельін құрастыру
Жаңашыл,креативті,педагог,шығармашыл,толерантты,медбике модельін құрастыру Меры по предотвращению распространения птичьего гриппа на территории Республики Башкортостан
Меры по предотвращению распространения птичьего гриппа на территории Республики Башкортостан Жұмыс қабілетінің жұмыс кезіңдегі өзгерістері. Белсенді демалу
Жұмыс қабілетінің жұмыс кезіңдегі өзгерістері. Белсенді демалу Медицина XXI века
Медицина XXI века КТ, МРТ, радіонуклідна діагностика молочної залози
КТ, МРТ, радіонуклідна діагностика молочної залози Исследование ассортимента и реализации комбинированных обезболивающих препаратов на основе метамизола натрия
Исследование ассортимента и реализации комбинированных обезболивающих препаратов на основе метамизола натрия Т-лимфоциты CD4+ и CD8+ в лимфоидных органах (иммунология, лекция 5)
Т-лимфоциты CD4+ и CD8+ в лимфоидных органах (иммунология, лекция 5) Расстройства личности
Расстройства личности Исследование метаболических эффектов льняного масла у крыс с интоксикацией тетрахлорметаном
Исследование метаболических эффектов льняного масла у крыс с интоксикацией тетрахлорметаном Аминогликозиды. Описание
Аминогликозиды. Описание Отделение онкореабилитации
Отделение онкореабилитации УМП Опухоли-2
УМП Опухоли-2 Эвтаназия. Философ Фрэнсис Бэкон
Эвтаназия. Философ Фрэнсис Бэкон Всё о Straumann. Расширенный курс для ортопедов и хирургов
Всё о Straumann. Расширенный курс для ортопедов и хирургов