- Построение QSAR модели для предсказания активности ингибиторов ренина

Содержание
- 2. ЦЕЛЬ И ЗАДАЧИ Цель: построить модель машинного обучения на основе алгоритма “случайных лесов” (random forest), которая
- 3. АКТУАЛЬНОСТЬ Сердечно-сосудистые заболевания являются основной причиной смерти и инвалидизации во всем мире. Поиск новых способов лечения
- 4. МАТЕРИАЛЫ И МЕТОДЫ Машинное обучение (machine learning, ML) – совокупность методов искусственного интеллекта, позволяющих строить алгоритмы
- 5. Скрипты для обработки данных и построения модели были написаны на языке программирования Python. Для 1D представления
- 6. Для отбора ингибиторов ренина использовалась база данных ChEMBL. Всего было найдено 5154 лиганда. Для дальнейшей работы
- 7. Фингерпринты – представление молекул в виде битовой строки, где каждый бит соответствует наличию (1) либо отсутствию
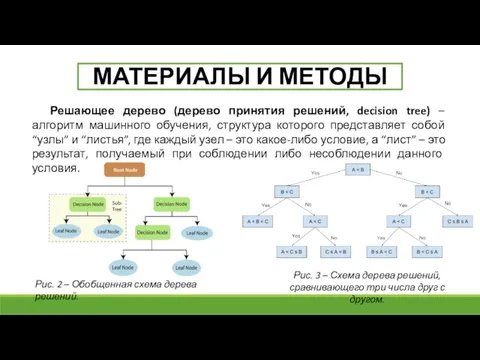
- 8. Решающее дерево (дерево принятия решений, decision tree) – алгоритм машинного обучения, структура которого представляет собой “узлы”
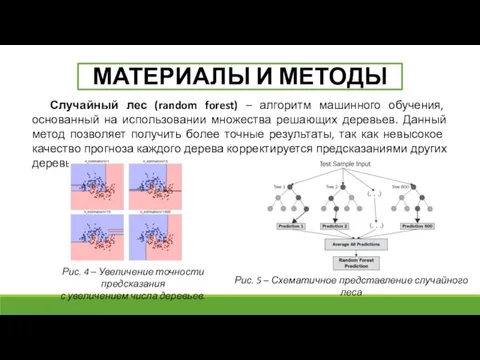
- 9. Случайный лес (random forest) – алгоритм машинного обучения, основанный на использовании множества решающих деревьев. Данный метод
- 10. МАТЕРИАЛЫ И МЕТОДЫ В качестве меры активности молекул использовалась IC50. Для удобства анализа данной меры активности
- 11. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ С помощью метода RandomForestClassifier была построена QSAR модель “случайных лесов”. Обучение модели
- 12. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ Метрики качества модели составили: Точность предсказания на тренировочных данных: 0,9472; Точность предсказания
- 13. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ Конфузионная матрица (confusion matrix) – таблица, представляющая результаты предсказания в сравнении с
- 14. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ По метрикам качества модели можно сделать вывод, что данная модель находит практически
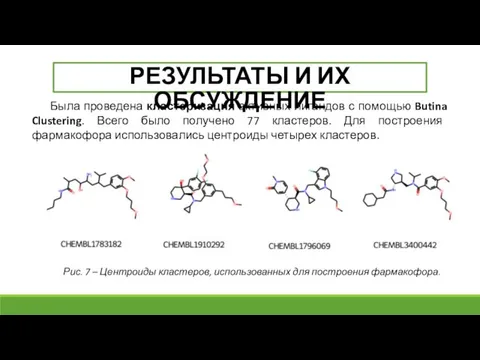
- 15. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ Была проведена кластеризация активных лигандов с помощью Butina Clustering. Всего было получено
- 16. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ Построение фармакофора проводилось с помощью алгоритма MAPex. Полученный фармакофор имеет 4 фармакофорных
- 17. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ По данному фармакофору был проведен поиск в базе данных ZINCPharmer, были сохранены
- 18. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ 1811 соединений, которые удовлетворяли критериям Липински были проверены по фильтру PAINS (Pan-assay
- 19. Для оставшихся 1245 соединений были получены их фингерпринты. С использованием QSAR модели, которая была построена ранее
- 21. Скачать презентацию

Слайд 3АКТУАЛЬНОСТЬ
Сердечно-сосудистые заболевания являются основной причиной смерти и инвалидизации во всем мире. Поиск
АКТУАЛЬНОСТЬ
Сердечно-сосудистые заболевания являются основной причиной смерти и инвалидизации во всем мире. Поиск

Слайд 4МАТЕРИАЛЫ И МЕТОДЫ
Машинное обучение (machine learning, ML) – совокупность методов искусственного интеллекта,
МАТЕРИАЛЫ И МЕТОДЫ
Машинное обучение (machine learning, ML) – совокупность методов искусственного интеллекта,

Слайд 5Скрипты для обработки данных и построения модели были написаны на языке программирования
Скрипты для обработки данных и построения модели были написаны на языке программирования

Слайд 6Для отбора ингибиторов ренина использовалась база данных ChEMBL. Всего было найдено 5154
Для отбора ингибиторов ренина использовалась база данных ChEMBL. Всего было найдено 5154

Слайд 7Фингерпринты – представление молекул в виде битовой строки, где каждый бит соответствует
Фингерпринты – представление молекул в виде битовой строки, где каждый бит соответствует

Слайд 8Решающее дерево (дерево принятия решений, decision tree) – алгоритм машинного обучения, структура
Решающее дерево (дерево принятия решений, decision tree) – алгоритм машинного обучения, структура
Слайд 9Случайный лес (random forest) – алгоритм машинного обучения, основанный на использовании множества
Случайный лес (random forest) – алгоритм машинного обучения, основанный на использовании множества
Слайд 10МАТЕРИАЛЫ И МЕТОДЫ
В качестве меры активности молекул использовалась IC50. Для удобства анализа
МАТЕРИАЛЫ И МЕТОДЫ
В качестве меры активности молекул использовалась IC50. Для удобства анализа

Слайд 11РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
С помощью метода RandomForestClassifier была построена QSAR модель “случайных
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
С помощью метода RandomForestClassifier была построена QSAR модель “случайных

Слайд 12РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Метрики качества модели составили:
Точность предсказания на тренировочных данных: 0,9472;
Точность
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Метрики качества модели составили:
Точность предсказания на тренировочных данных: 0,9472;
Точность

Слайд 13РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Конфузионная матрица (confusion matrix) – таблица, представляющая результаты предсказания
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Конфузионная матрица (confusion matrix) – таблица, представляющая результаты предсказания

Слайд 14РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
По метрикам качества модели можно сделать вывод, что данная
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
По метрикам качества модели можно сделать вывод, что данная

Слайд 15РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Была проведена кластеризация активных лигандов с помощью Butina Clustering.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Была проведена кластеризация активных лигандов с помощью Butina Clustering.
Слайд 16РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Построение фармакофора проводилось с помощью алгоритма MAPex. Полученный фармакофор
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Построение фармакофора проводилось с помощью алгоритма MAPex. Полученный фармакофор

Слайд 17РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
По данному фармакофору был проведен поиск в базе данных
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
По данному фармакофору был проведен поиск в базе данных

Слайд 18РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
1811 соединений, которые удовлетворяли критериям Липински были проверены по
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
1811 соединений, которые удовлетворяли критериям Липински были проверены по

Слайд 19Для оставшихся 1245 соединений были получены их фингерпринты. С использованием QSAR модели,
Для оставшихся 1245 соединений были получены их фингерпринты. С использованием QSAR модели,

 Фармакоэпидемиологический и фармакоэкономический анализ лекарственных назначений в реабилитационном периоде инфаркта миокарда
Фармакоэпидемиологический и фармакоэкономический анализ лекарственных назначений в реабилитационном периоде инфаркта миокарда ТОВ Медична лабораторія
ТОВ Медична лабораторія Заболевания ИППП
Заболевания ИППП История обучения первой помощи на Среднем Урале
История обучения первой помощи на Среднем Урале Применение здоовьесберегающих технологий для работоспособности учащихся с ограниченными возможностями здоровья
Применение здоовьесберегающих технологий для работоспособности учащихся с ограниченными возможностями здоровья Обоснование необходимости системы Медицинская Сигнализация SOS
Обоснование необходимости системы Медицинская Сигнализация SOS Хранение лекарственных средств и средства бытовой химии
Хранение лекарственных средств и средства бытовой химии Уход за кожей и естественными складками, умывание тяжелобольного пациента
Уход за кожей и естественными складками, умывание тяжелобольного пациента Рак легкого (Лекция №9)
Рак легкого (Лекция №9) :Балалардағы бауыр, өт қабы және ұйқы безінің сәулелік диагностикасы
:Балалардағы бауыр, өт қабы және ұйқы безінің сәулелік диагностикасы Хирургический шов
Хирургический шов Патология кровообращения
Патология кровообращения Философия врачевания
Философия врачевания Эмоционально-волевые нарушения. Депрессивные состояния
Эмоционально-волевые нарушения. Депрессивные состояния Chronic gastritis
Chronic gastritis Учебная медицинская карта пациента
Учебная медицинская карта пациента Ведение больных с ювенильным маточным кровотечением
Ведение больных с ювенильным маточным кровотечением Ветсанэкспертиза туш и органов животных, пораженных биологическими средствами
Ветсанэкспертиза туш и органов животных, пораженных биологическими средствами Аппендикулярный инфильтрат
Аппендикулярный инфильтрат Вебинар. Умный фитнес. Модуль 1. Диагностика: диафрагма, тазовое дно, мышцы кора
Вебинар. Умный фитнес. Модуль 1. Диагностика: диафрагма, тазовое дно, мышцы кора Закаливающие водные процедуры
Закаливающие водные процедуры Жұқпалы ауруларды тіркеу-есеп медициналық құжат
Жұқпалы ауруларды тіркеу-есеп медициналық құжат Менингококовые инфекции
Менингококовые инфекции Иммунное реагирование при различных инфекциях
Иммунное реагирование при различных инфекциях Дневник питания и тренировок на 14 дней
Дневник питания и тренировок на 14 дней Новообразования, их медико-социальная значимость
Новообразования, их медико-социальная значимость Игровые приёмы, применяемые при коррекции оптической дисграфии у детей с ЗПР
Игровые приёмы, применяемые при коррекции оптической дисграфии у детей с ЗПР Лікарські засоби, що діють переважно на Центральну нервову систему (ЦНС)
Лікарські засоби, що діють переважно на Центральну нервову систему (ЦНС)