- Наследственные заболевания. Лекция 2. Лизосомные болезни накопления

Содержание
- 3. ЭУКАРИОТИЧЕСКАЯ КЛЕТКА
- 5. Лизосомные болезни накопления Концепция лизосомных болезней накопления сложилась в результате изучения гликогеноза II типа (Помпе). Факт
- 6. Мукополисахаридозы (МПС) Недостаточность лизосомальных ферментов изменяет катаболизм гликозаминогликанов (ГАГ) с накоплением их в лизосомах и приводит
- 7. Мукополисахаридоз 1 тип синдром Гурлер Выраженная умственная отсталость Черепно-лицевые дизморфии (грубые черты лица) Помутнение роговицы Гепатоспленомегалия
- 8. МПС I тип Гурлер
- 9. МПС I тип Гурлера (внутренняя гидроцефалия)
- 10. МПС IS тип синдром Гурлер-Шейе доброкачественное течение сохранный интеллект тугоподвижность суставов
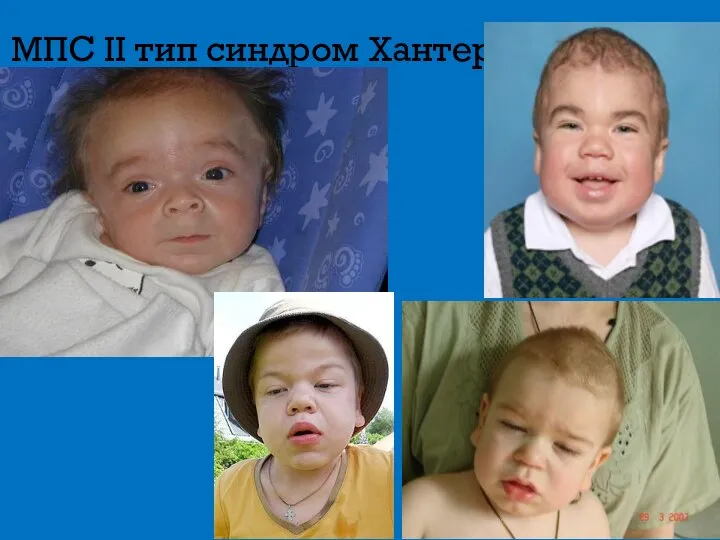
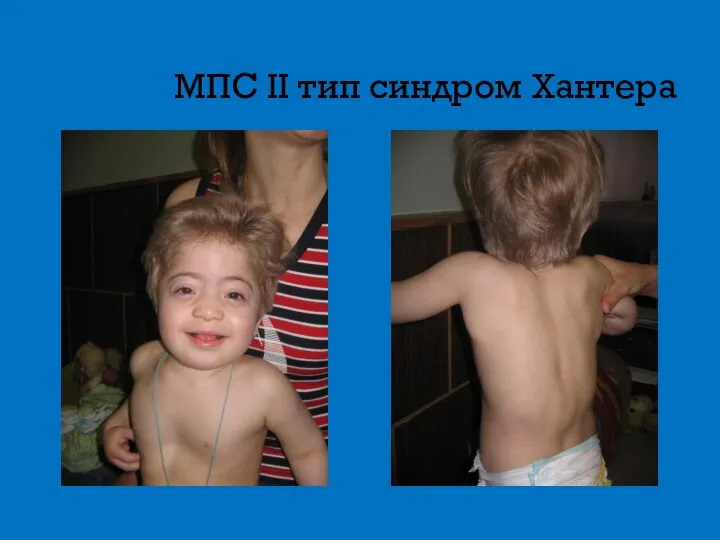
- 11. МПС II тип синдром Хантера Доброкачественное течение Паховые и пупочные грыжи Шумное дыхание Тугоподвижность суставов Задержка
- 12. МПС II тип синдром Хантера
- 13. МПС II тип синдром Хантера
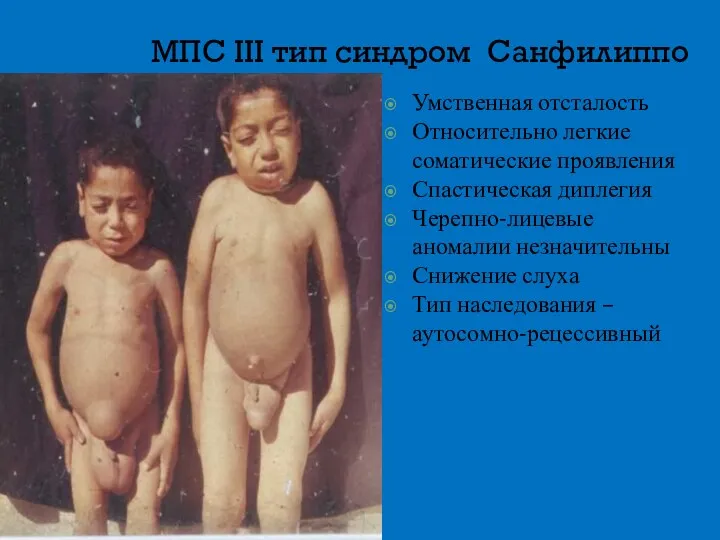
- 14. МПС III тип синдром Санфилиппо Умственная отсталость Относительно легкие соматические проявления Спастическая диплегия Черепно-лицевые аномалии незначительны
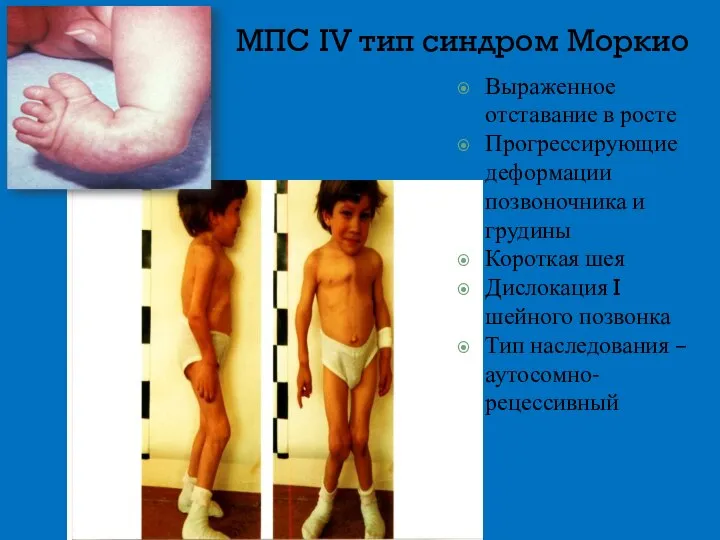
- 15. МПС IV тип синдром Моркио Выраженное отставание в росте Прогрессирующие деформации позвоночника и грудины Короткая шея
- 16. МПС VI тип синдром Марото-Лами Сохранный интеллект Помутнение роговицы Снижение слуха Тугоподвижность суставов Низкий рост Гепатоспленомегалия
- 17. муколипидоз Грубые черты лица Скелетные деформации Симптом «вишневой косточки» на глазном дне Поражение ЦНС (судороги, умственная
- 18. маннозидоз Грубые черты лица Макроглоссия Скелетные аномалии Большой живот Пупочная грыжа Нейросенсорная глухота
- 19. Болезнь Помпе Впервые описана в 1932г голландским патологом J.C. Pompe Недостаточность фермента кислой α-глюкозидазы (GAA) Более
- 20. Орган - мишень Поражение мышечной ткани – накопление гликогена в лизосомах миоцитов ↓ Прогрессивное нарушение функции
- 21. Клиническая картина Форма с ранним началом (младенческая) 1:138 000 начало до 12мес Быстрое прогрессирование 2 подтипа:
- 22. Клиническая картина Форма с поздним началом 1:57 000 Манифестация после 12мес Медленное прогрессирование Миопатия Вариабельность симптомов
- 23. Мышечная система Инфантильная форма Прогрессирующая мышечная слабость, тяжелая гипотония, нарушение двигательной активности, задержка моторного развития, миопатическое
- 24. Дыхательная и сердечно-сосудистая системы Прогрессирующая дыхательная недостаточность Частые ОРВИ / аспирационные пневмонии Прогрессирующая кардиомиопатия Кардиомегалия Сердечная
- 25. Желудочно-кишечный тракт Сложности при кормлении Макроглоссия Задержка роста и развития Сложно поддерживать нормальный вес (склонность к
- 26. Диагностика Осмотр Пальпация мышц Упражнения на движения ЭМГ Биопсия мышц Определение активности фермента в фибробластах кожи,
- 28. Лечение С начала 2006г появился препарат для фермент заместительной терапии – Миозим (Альглюкозидаза альфа) Производитель:Genzyme Europe
- 29. Результаты лечения После 3-х инфузий возможность самостоятельного дыхания (перевод с ИВЛ) Аппарат ИВЛ может использоваться как
- 30. Ранняя детская амавротическая идиотия Болезнь Тея-Сакса GM2-ганглиозидоз Названа в честь британского офтальмолога Уоррена Тея обнаружившего красное
- 31. Warren Tay (1843—1927) Bernard Sachs (1858—1944) Болезнь распространена в еврейских семьях Восточной Европы. В очень раннем
- 32. «Синдром вишневой косточки» - характерный диагностический признак заболевания при офтальмоскопическом исследовании
- 33. Ганглиозиды - это сложные гликолипиды (комплексы сахаров и липидов). В клетках ганглиозиды разрушаются и собираются вновь
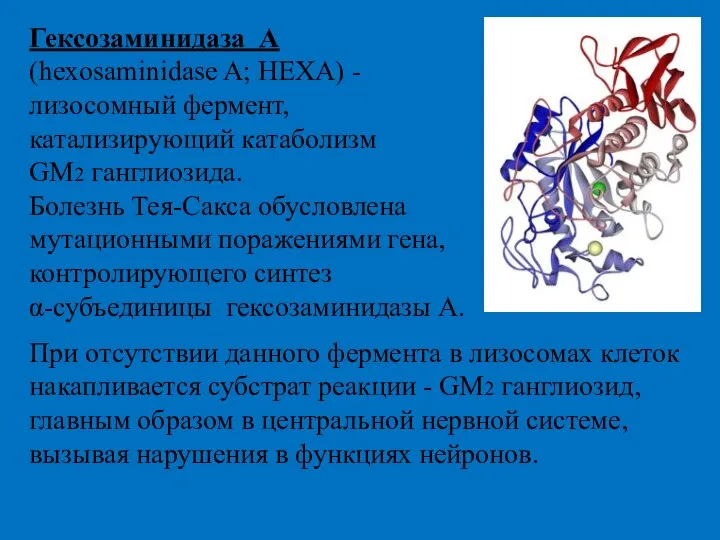
- 34. Гексозаминидаза А (hexosaminidase A; HEXA) - лизосомный фермент, катализирующий катаболизм GM2 ганглиозида. Болезнь Тея-Сакса обусловлена мутационными
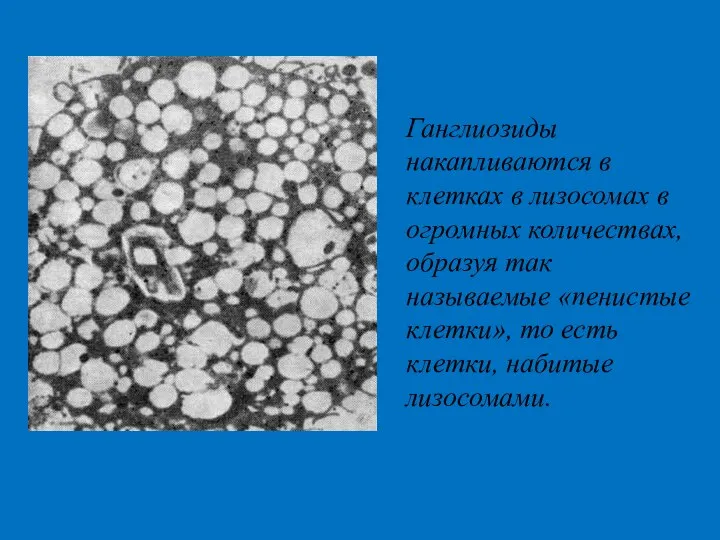
- 35. Ганглиозиды накапливаются в клетках в лизосомах в огромных количествах, образуя так называемые «пенистые клетки», то есть
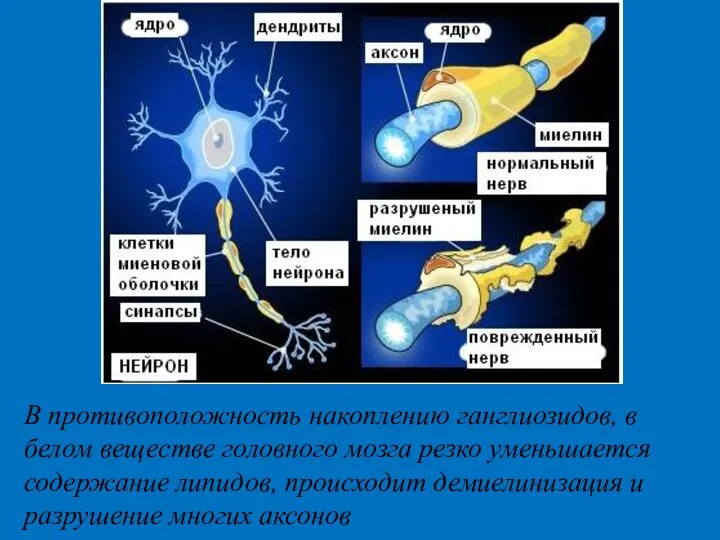
- 36. В противоположность накоплению ганглиозидов, в белом веществе головного мозга резко уменьшается содержание липидов, происходит демиелинизация и
- 37. Дети с болезнью Тея-Сакса
- 38. Частота встречаемости заболевания в среднем 1 случай на 250 000 – 500 000 человек, Среди некоторых
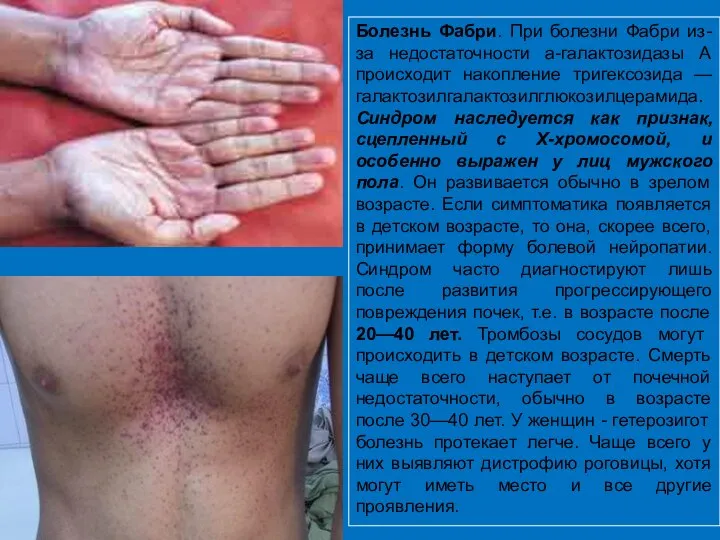
- 39. Болезнь Фабри. При болезни Фабри из-за недостаточности а-галактозидазы А происходит накопление тригексозида — галактозилгалактозилглюкозилцерамида. Синдром наследуется
- 41. Галактозилцерамидный липидоз Краббе или шаровидно-клеточная лейкодистрофия, проявляется в младенчестве из-за недостаточности галактозилцерамид-b-галактозидазы. Для него типичны начало
- 42. Митохондриальные заболевания
- 43. Митохондриальные заболевания — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических
- 44. Можно выделить две группы митохондриальных заболеваний: • Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за
- 45. Наследование митохондриальных болезней Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно
- 46. Типы заболеваний Помимо относительно распространённой митохондриальной миопатии, встречаются: митохондриальный сахарный диабет, сопровождающийся глухотой (DAD, MIDD, синдром
- 47. синдром Лея или подострая некротизирующая энцефаломиопатия : после начального нормального постнатального развития болезнь проявляется обычно в
- 48. синдром Вольфа-Паркинсона-Уайта
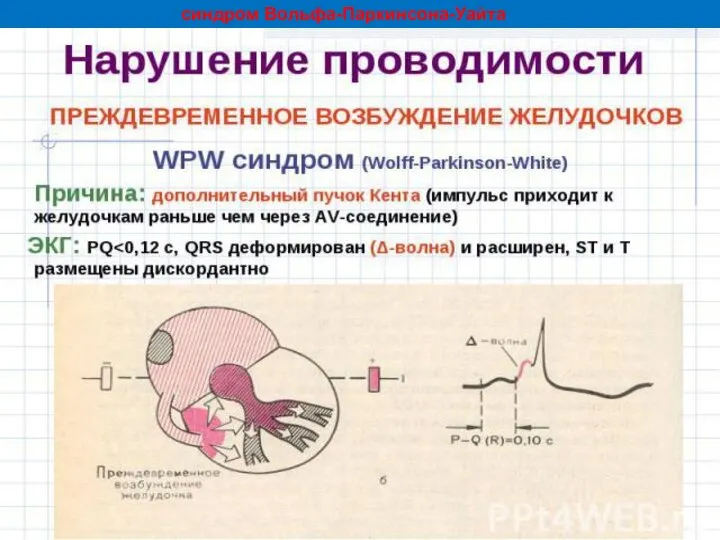
- 49. Синдром Вольфа-Паркинсона-Уайта — наиболее частый синдром преждевременного возбуждения желудочков (его наблюдают у 0,1 — 0,3 %
- 50. Лечение Существует несколько способов лечения синдрома WPW: Антиаритмическая терапия — при постоянном приеме медикаментозных препаратов. Важно:
- 52. Скачать презентацию
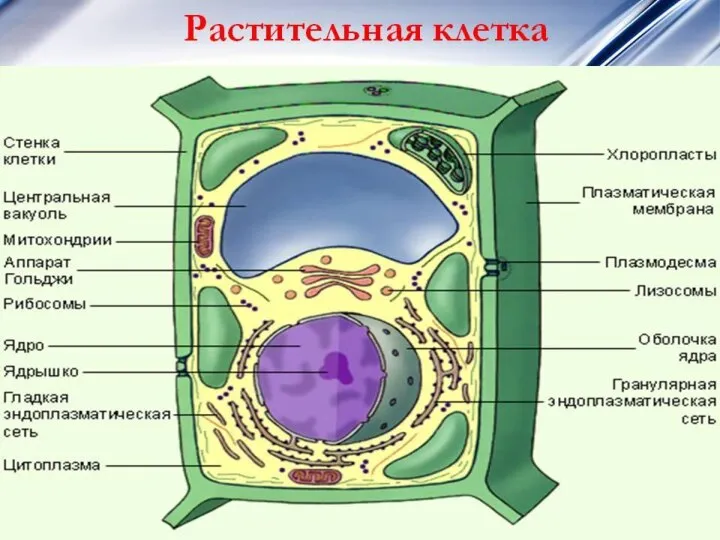
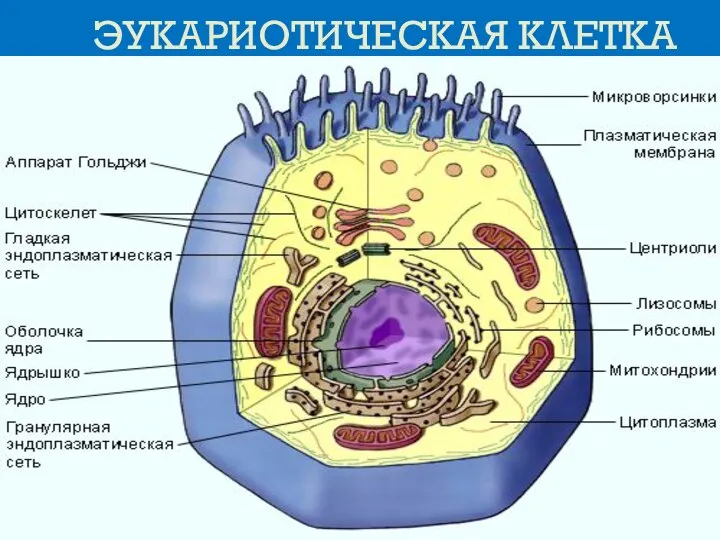
Слайд 3ЭУКАРИОТИЧЕСКАЯ КЛЕТКА
ЭУКАРИОТИЧЕСКАЯ КЛЕТКА
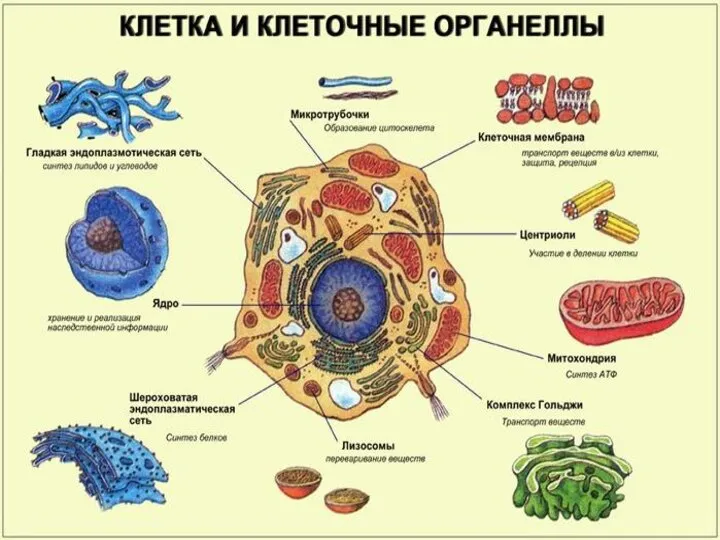
Слайд 5Лизосомные болезни накопления
Концепция лизосомных болезней накопления сложилась в результате изучения гликогеноза II
Лизосомные болезни накопления
Концепция лизосомных болезней накопления сложилась в результате изучения гликогеноза II

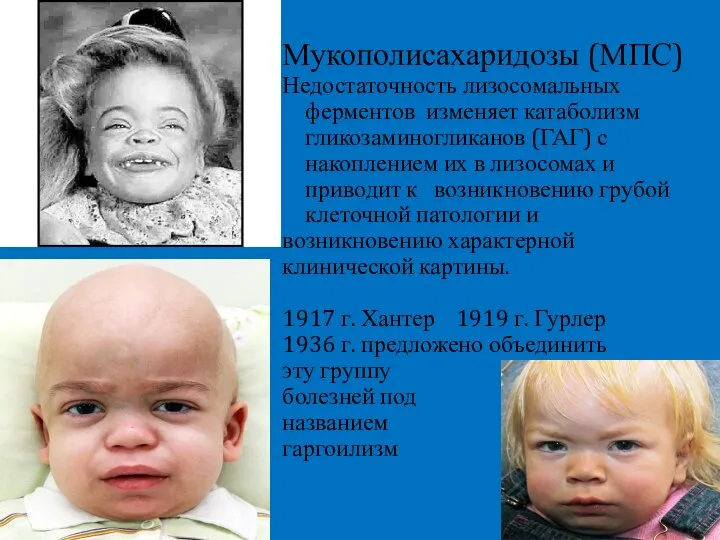
Слайд 6Мукополисахаридозы (МПС)
Недостаточность лизосомальных ферментов изменяет катаболизм гликозаминогликанов (ГАГ) с накоплением их в
Недостаточность лизосомальных ферментов изменяет катаболизм гликозаминогликанов (ГАГ) с накоплением их в
Слайд 7Мукополисахаридоз 1 тип синдром Гурлер
Выраженная умственная отсталость
Черепно-лицевые дизморфии (грубые черты лица)
Помутнение роговицы
Гепатоспленомегалия
Грубые
Мукополисахаридоз 1 тип синдром Гурлер
Выраженная умственная отсталость
Черепно-лицевые дизморфии (грубые черты лица)
Помутнение роговицы
Гепатоспленомегалия
Грубые

Слайд 8МПС I тип Гурлер
МПС I тип Гурлер
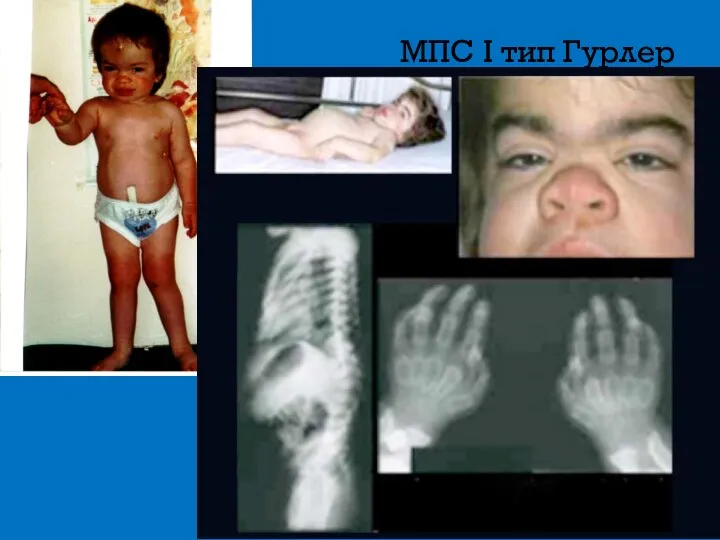
Слайд 9МПС I тип Гурлера
(внутренняя гидроцефалия)
МПС I тип Гурлера
(внутренняя гидроцефалия)
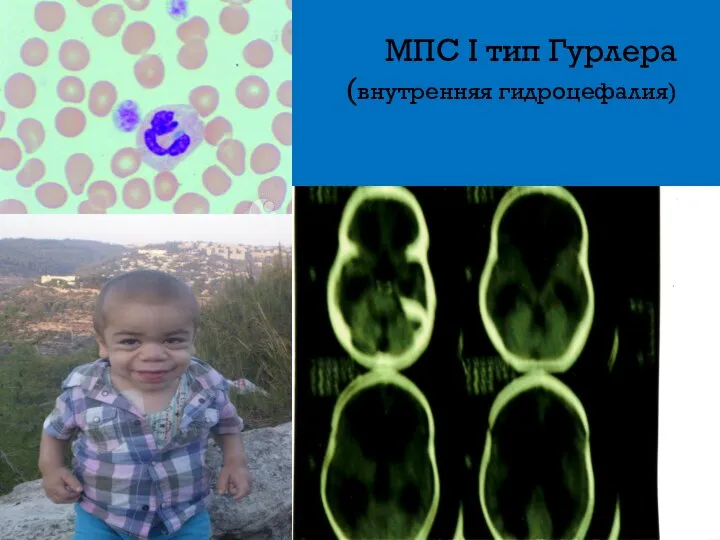
Слайд 10МПС IS тип синдром Гурлер-Шейе
доброкачественное течение
сохранный интеллект
тугоподвижность суставов
МПС IS тип синдром Гурлер-Шейе
доброкачественное течение
сохранный интеллект
тугоподвижность суставов
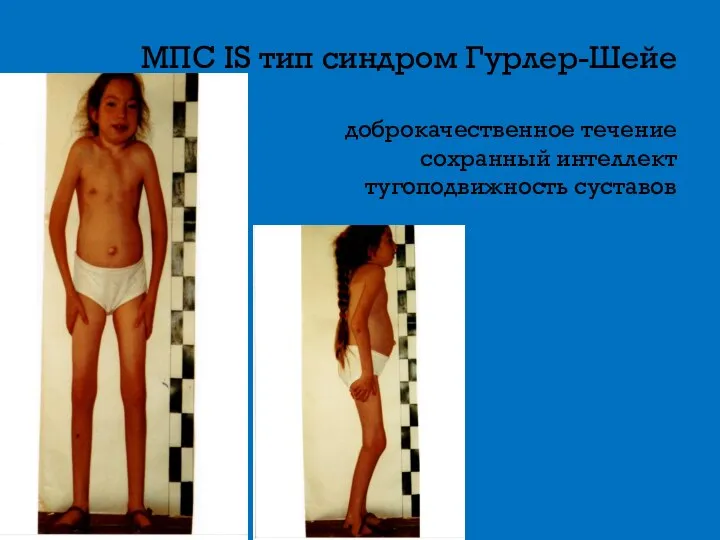
Слайд 11МПС II тип синдром Хантера
Доброкачественное течение
Паховые и пупочные грыжи
Шумное дыхание
Тугоподвижность суставов
Задержка роста
Гепатоспленомегалия
Тип
МПС II тип синдром Хантера
Доброкачественное течение
Паховые и пупочные грыжи
Шумное дыхание
Тугоподвижность суставов
Задержка роста
Гепатоспленомегалия
Тип
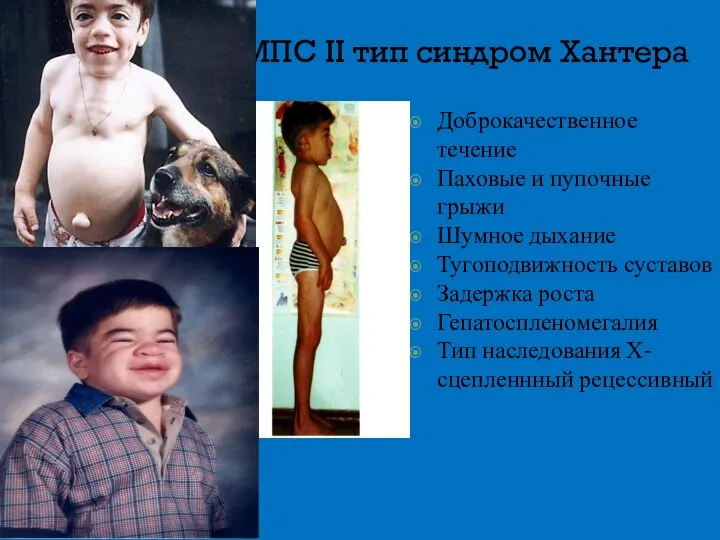
Слайд 12МПС II тип синдром Хантера
МПС II тип синдром Хантера
Слайд 13МПС II тип синдром Хантера
МПС II тип синдром Хантера
Слайд 14МПС III тип синдром Санфилиппо
Умственная отсталость
Относительно легкие соматические проявления
Спастическая диплегия
Черепно-лицевые аномалии незначительны
Снижение
МПС III тип синдром Санфилиппо
Умственная отсталость
Относительно легкие соматические проявления
Спастическая диплегия
Черепно-лицевые аномалии незначительны
Снижение
Слайд 15МПС IV тип синдром Моркио
Выраженное отставание в росте
Прогрессирующие деформации позвоночника и грудины
Короткая
МПС IV тип синдром Моркио
Выраженное отставание в росте
Прогрессирующие деформации позвоночника и грудины
Короткая
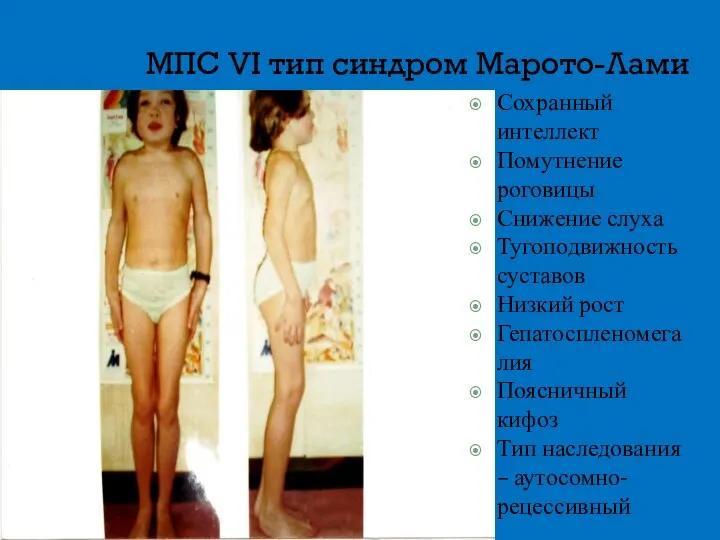
Слайд 16МПС VI тип синдром Марото-Лами
Сохранный интеллект
Помутнение роговицы
Снижение слуха
Тугоподвижность суставов
Низкий рост
Гепатоспленомегалия
Поясничный кифоз
Тип наследования
МПС VI тип синдром Марото-Лами
Сохранный интеллект
Помутнение роговицы
Снижение слуха
Тугоподвижность суставов
Низкий рост
Гепатоспленомегалия
Поясничный кифоз
Тип наследования
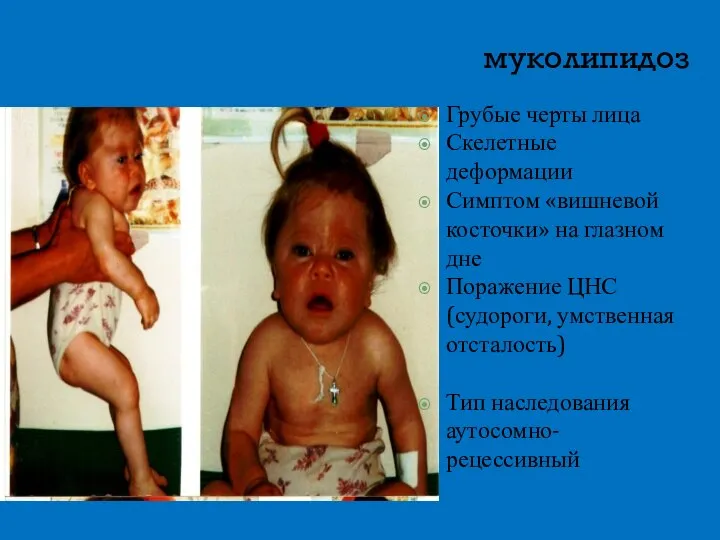
Слайд 17муколипидоз
Грубые черты лица
Скелетные деформации
Симптом «вишневой косточки» на глазном дне
Поражение ЦНС (судороги, умственная
муколипидоз
Грубые черты лица
Скелетные деформации
Симптом «вишневой косточки» на глазном дне
Поражение ЦНС (судороги, умственная
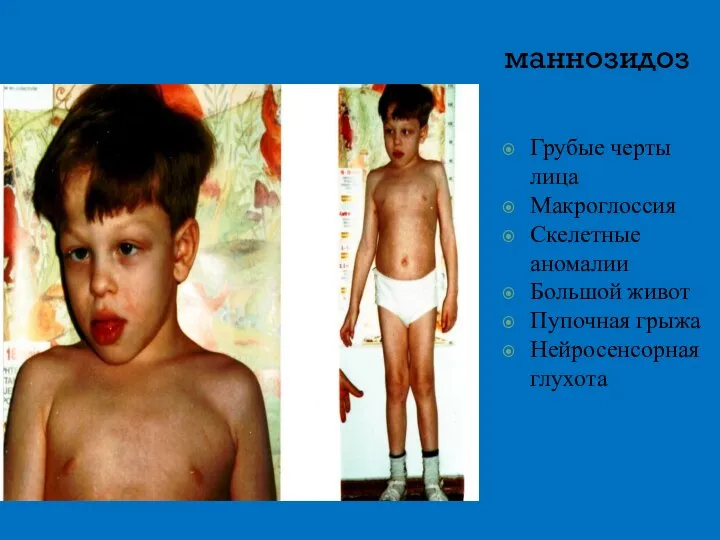
Слайд 18маннозидоз
Грубые черты лица
Макроглоссия
Скелетные аномалии
Большой живот
Пупочная грыжа
Нейросенсорная глухота
маннозидоз
Грубые черты лица
Макроглоссия
Скелетные аномалии
Большой живот
Пупочная грыжа
Нейросенсорная глухота
Слайд 19Болезнь Помпе
Впервые описана в 1932г голландским патологом J.C. Pompe
Недостаточность фермента кислой α-глюкозидазы
Болезнь Помпе
Впервые описана в 1932г голландским патологом J.C. Pompe
Недостаточность фермента кислой α-глюкозидазы

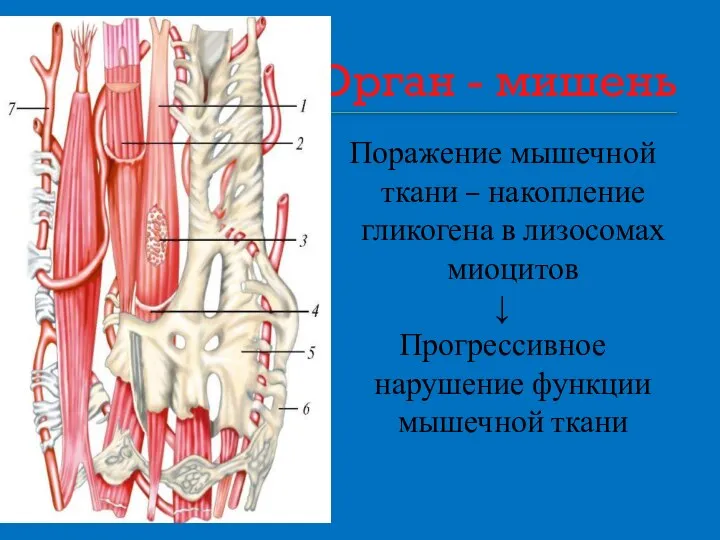
Слайд 20Орган - мишень
Поражение мышечной ткани – накопление гликогена в лизосомах миоцитов
↓
Прогрессивное нарушение
Орган - мишень
Поражение мышечной ткани – накопление гликогена в лизосомах миоцитов
↓
Прогрессивное нарушение
Слайд 21Клиническая картина
Форма с ранним началом (младенческая) 1:138 000
начало до 12мес
Быстрое прогрессирование
2 подтипа:
Клиническая картина
Форма с ранним началом (младенческая) 1:138 000
начало до 12мес
Быстрое прогрессирование
2 подтипа:

Слайд 22Клиническая картина
Форма с поздним началом 1:57 000
Манифестация после 12мес
Медленное прогрессирование
Миопатия
Вариабельность симптомов поражения
Клиническая картина
Форма с поздним началом 1:57 000
Манифестация после 12мес
Медленное прогрессирование
Миопатия
Вариабельность симптомов поражения

Слайд 23Мышечная система
Инфантильная форма
Прогрессирующая мышечная слабость, тяжелая гипотония, нарушение двигательной активности, задержка моторного
Мышечная система
Инфантильная форма
Прогрессирующая мышечная слабость, тяжелая гипотония, нарушение двигательной активности, задержка моторного

Слайд 24Дыхательная и сердечно-сосудистая системы
Прогрессирующая дыхательная недостаточность
Частые ОРВИ / аспирационные пневмонии
Прогрессирующая кардиомиопатия
Кардиомегалия
Сердечная недостаточность
Летальный
Дыхательная и сердечно-сосудистая системы
Прогрессирующая дыхательная недостаточность
Частые ОРВИ / аспирационные пневмонии
Прогрессирующая кардиомиопатия
Кардиомегалия
Сердечная недостаточность
Летальный

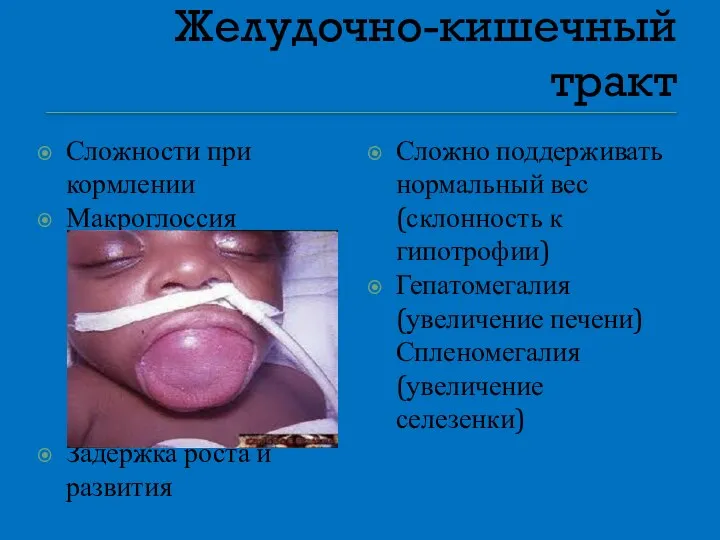
Слайд 25Желудочно-кишечный тракт
Сложности при кормлении
Макроглоссия
Задержка роста и развития
Сложно поддерживать нормальный вес (склонность к
Желудочно-кишечный тракт
Сложности при кормлении
Макроглоссия
Задержка роста и развития
Сложно поддерживать нормальный вес (склонность к
Слайд 26Диагностика
Осмотр
Пальпация мышц
Упражнения на движения
ЭМГ
Биопсия мышц
Определение активности фермента в фибробластах кожи, лимфоцитах, миоцитах,
Диагностика
Осмотр
Пальпация мышц
Упражнения на движения
ЭМГ
Биопсия мышц
Определение активности фермента в фибробластах кожи, лимфоцитах, миоцитах,


Слайд 28Лечение
С начала 2006г появился препарат для фермент заместительной терапии – Миозим (Альглюкозидаза
Лечение
С начала 2006г появился препарат для фермент заместительной терапии – Миозим (Альглюкозидаза

Слайд 29Результаты лечения
После 3-х инфузий возможность самостоятельного дыхания (перевод с ИВЛ) Аппарат ИВЛ может использоваться
Результаты лечения
После 3-х инфузий возможность самостоятельного дыхания (перевод с ИВЛ) Аппарат ИВЛ может использоваться

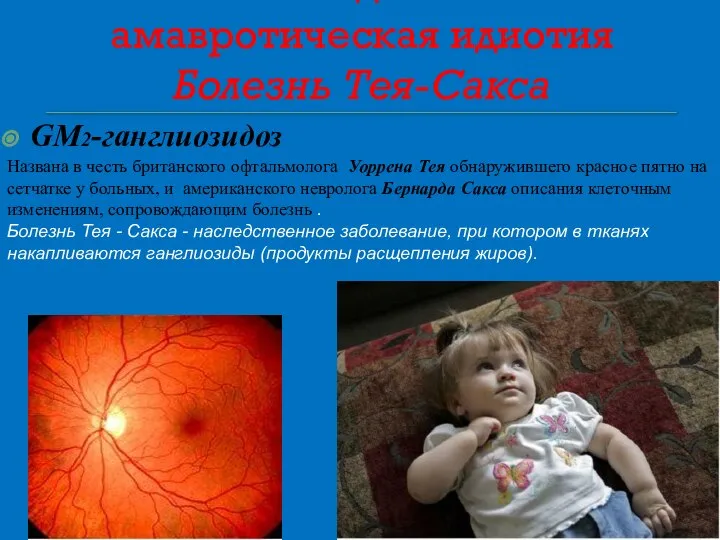
Слайд 30Ранняя детская амавротическая идиотия Болезнь Тея-Сакса
GM2-ганглиозидоз
Названа в честь британского офтальмолога Уоррена Тея
Ранняя детская амавротическая идиотия Болезнь Тея-Сакса
GM2-ганглиозидоз
Названа в честь британского офтальмолога Уоррена Тея
Слайд 31Warren Tay (1843—1927)
Bernard Sachs (1858—1944)
Болезнь распространена в еврейских семьях Восточной Европы. В
Warren Tay (1843—1927)
Bernard Sachs (1858—1944)
Болезнь распространена в еврейских семьях Восточной Европы. В

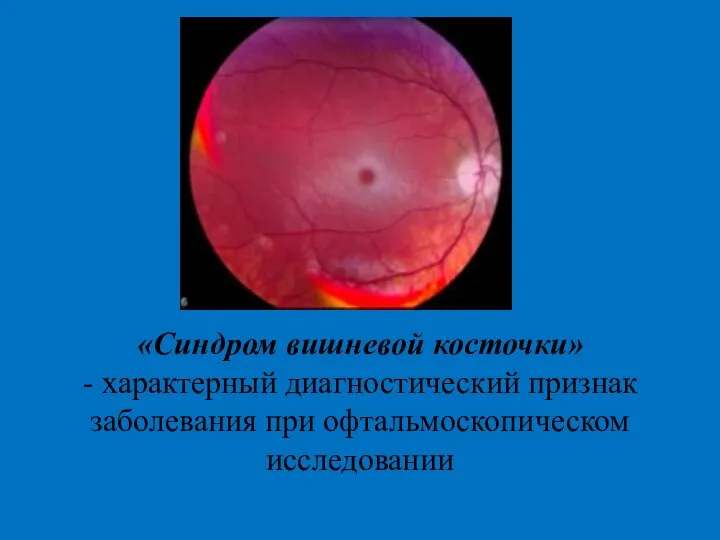
Слайд 32«Синдром вишневой косточки»
- характерный диагностический признак заболевания при офтальмоскопическом исследовании
«Синдром вишневой косточки»
- характерный диагностический признак заболевания при офтальмоскопическом исследовании
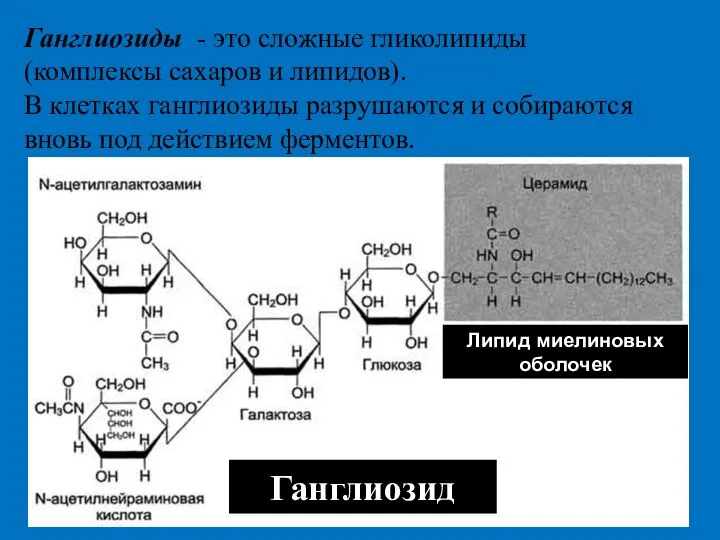
Слайд 33Ганглиозиды - это сложные гликолипиды
(комплексы сахаров и липидов).
В клетках ганглиозиды разрушаются и
Ганглиозиды - это сложные гликолипиды
(комплексы сахаров и липидов).
В клетках ганглиозиды разрушаются и
Слайд 34Гексозаминидаза А
(hexosaminidase A; HEXA) - лизосомный фермент,
катализирующий катаболизм
GM2 ганглиозида.
Гексозаминидаза А
(hexosaminidase A; HEXA) - лизосомный фермент,
катализирующий катаболизм
GM2 ганглиозида.
Слайд 35Ганглиозиды накапливаются в клетках в лизосомах в огромных количествах, образуя так называемые
Ганглиозиды накапливаются в клетках в лизосомах в огромных количествах, образуя так называемые
Слайд 36В противоположность накоплению ганглиозидов, в белом веществе головного мозга резко уменьшается содержание
В противоположность накоплению ганглиозидов, в белом веществе головного мозга резко уменьшается содержание
Слайд 37Дети с болезнью Тея-Сакса
Дети с болезнью Тея-Сакса

Слайд 38Частота встречаемости заболевания
в среднем 1 случай на 250 000 – 500 000
Частота встречаемости заболевания
в среднем 1 случай на 250 000 – 500 000

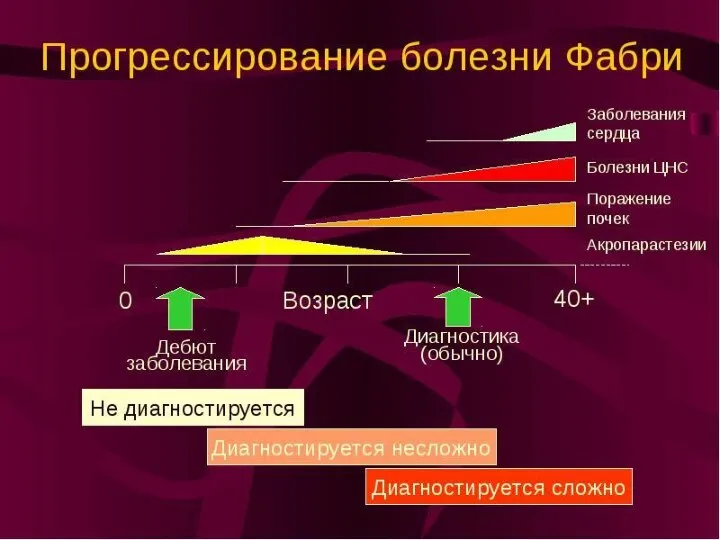
Слайд 39Болезнь Фабри. При болезни Фабри из-за недостаточности а-галактозидазы А происходит накопление тригексозида
Болезнь Фабри. При болезни Фабри из-за недостаточности а-галактозидазы А происходит накопление тригексозида
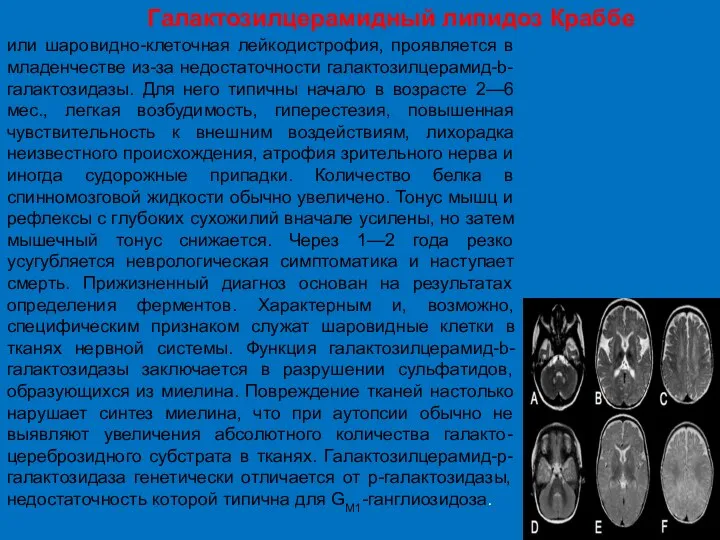
Слайд 41Галактозилцерамидный липидоз Краббе
или шаровидно-клеточная лейкодистрофия, проявляется в младенчестве из-за недостаточности галактозилцерамид-b-галактозидазы. Для
Галактозилцерамидный липидоз Краббе
или шаровидно-клеточная лейкодистрофия, проявляется в младенчестве из-за недостаточности галактозилцерамид-b-галактозидазы. Для
Слайд 42Митохондриальные заболевания
Митохондриальные заболевания
Слайд 43Митохондриальные заболевания — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими
Митохондриальные заболевания — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими
Слайд 44Можно выделить две группы митохондриальных заболеваний:
• Ярко выраженные наследственные синдромы, обусловленные мутациями генов,
Можно выделить две группы митохондриальных заболеваний:
• Ярко выраженные наследственные синдромы, обусловленные мутациями генов,

Слайд 45Наследование митохондриальных болезней
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой
Наследование митохондриальных болезней
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой

Слайд 46Типы заболеваний
Помимо относительно распространённой митохондриальной миопатии, встречаются:
митохондриальный сахарный диабет, сопровождающийся глухотой
Типы заболеваний
Помимо относительно распространённой митохондриальной миопатии, встречаются:
митохондриальный сахарный диабет, сопровождающийся глухотой

Слайд 47 синдром Лея или подострая некротизирующая энцефаломиопатия : после начального нормального постнатального
синдром Лея или подострая некротизирующая энцефаломиопатия : после начального нормального постнатального
Слайд 48синдром Вольфа-Паркинсона-Уайта
синдром Вольфа-Паркинсона-Уайта
Слайд 49Синдром Вольфа-Паркинсона-Уайта — наиболее частый синдром преждевременного возбуждения желудочков (его наблюдают у
Синдром Вольфа-Паркинсона-Уайта — наиболее частый синдром преждевременного возбуждения желудочков (его наблюдают у

Слайд 50Лечение
Существует несколько способов лечения синдрома WPW:
Антиаритмическая терапия — при постоянном приеме медикаментозных
Лечение
Существует несколько способов лечения синдрома WPW:
Антиаритмическая терапия — при постоянном приеме медикаментозных

 Пріонні захворювання. Трансмісивні губчасті підгостріенцефалопатії
Пріонні захворювання. Трансмісивні губчасті підгостріенцефалопатії Понятие риска и общие аспекты использования методологии риска в системах СГМ и управления санитарно-эпидемиологической ситуацией
Понятие риска и общие аспекты использования методологии риска в системах СГМ и управления санитарно-эпидемиологической ситуацией Минералокортикоиды (ДОКСА): влияние на минеральный обмен, показания к применению
Минералокортикоиды (ДОКСА): влияние на минеральный обмен, показания к применению Опухоли женских половых органов
Опухоли женских половых органов Тромболитическая терапия при тромбоэмболии лёгочной артерии (ТЭЛА)
Тромболитическая терапия при тромбоэмболии лёгочной артерии (ТЭЛА) Анестезия при кесаревом сечении
Анестезия при кесаревом сечении Замена воска на пластмассу
Замена воска на пластмассу ЭС 2 БАЗА
ЭС 2 БАЗА ЭКГ при ишемической болезни сердца
ЭКГ при ишемической болезни сердца Сравнение состояния опорнодвигательного аппарата при гиподинамии и в условиях физической нагрузки
Сравнение состояния опорнодвигательного аппарата при гиподинамии и в условиях физической нагрузки АІТ жоғарғы бөлігінің қан кетуінде көрсетілетін шұғыл көмек
АІТ жоғарғы бөлігінің қан кетуінде көрсетілетін шұғыл көмек Рентгенологические синдромы заболеваний легких
Рентгенологические синдромы заболеваний легких Chronic gastritis
Chronic gastritis Антисептика. Определение. Значение работ Пастера, Листера, Пирогова. Виды антисептики и современные антисептические вещества
Антисептика. Определение. Значение работ Пастера, Листера, Пирогова. Виды антисептики и современные антисептические вещества Развитие и образование детей со сложными нарушениями развития
Развитие и образование детей со сложными нарушениями развития Одышка: клинические проявления
Одышка: клинические проявления Изменения слизистой оболочки полости рта при заболеваниях сердечно-сосудистой системы
Изменения слизистой оболочки полости рта при заболеваниях сердечно-сосудистой системы Грипп Грипп А/H1N1/ Калифорния/04/09/ Грипп,вызванный новым пандемическим вирусом A/H1N1swl/ доц. Кулагина М.Г.
Грипп Грипп А/H1N1/ Калифорния/04/09/ Грипп,вызванный новым пандемическим вирусом A/H1N1swl/ доц. Кулагина М.Г. Лечебно–диагностические вмешательства и сестринский уход при заболеваниях уха и сосцевидного отростка
Лечебно–диагностические вмешательства и сестринский уход при заболеваниях уха и сосцевидного отростка Кохлеарная имплантация
Кохлеарная имплантация Основы физиотерапии
Основы физиотерапии Вегето-сосудистая дистония. Лекция 24
Вегето-сосудистая дистония. Лекция 24 Биоэтика. Лекция 1
Биоэтика. Лекция 1 Перемещение больного
Перемещение больного Правила работы в микробиологической лаборатории
Правила работы в микробиологической лаборатории Департаменты медико-информационного анализа
Департаменты медико-информационного анализа Философия и медицина
Философия и медицина Сегментарно-рефлекторный массаж
Сегментарно-рефлекторный массаж