- Мукополисахаридозы

Содержание
- 2. Мукополисахаридозы (мукополисахариды + -ōsis) группа наследственных болезней соединительной ткани, обусловленных нарушением обмена гликозаминогликанов (кислых мукополисахаридов) в
- 3. Мукополисахаридоз типа I-Н (синдром Гурлер). Впервые описан немецким педиатром Гурлер (G. Hurler) в 1919 г. Часто
- 4. Рис. 1. Синдром Гурлер: типичные внешние проявления.
- 5. Аномалии лицевого скелета при синдроме Hurler
- 6. Помутнение роговицы при синдроме Hurler Мукополисахаридозы Группа наследственных заболеваний, обусловленных дефицитом ферментов гидролиза мукополисахаридов (глюкозидазы). Продукты
- 7. При рентгенологическом исследовании (рис. 2) выявляются характерные изменения позвонков и позвоночного столба (тела позвонков кубовидные с
- 8. Рис. 2а). Рентгенологические признаки синдрома Гурлер — изменения ребер и позвоночника.
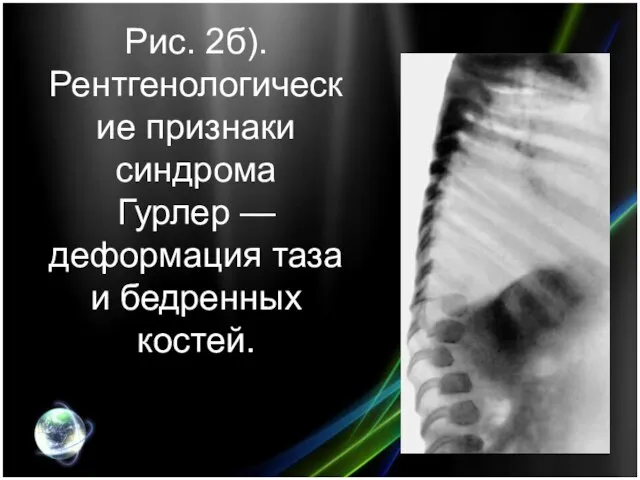
- 9. Рис. 2б). Рентгенологические признаки синдрома Гурлер — деформация таза и бедренных костей.
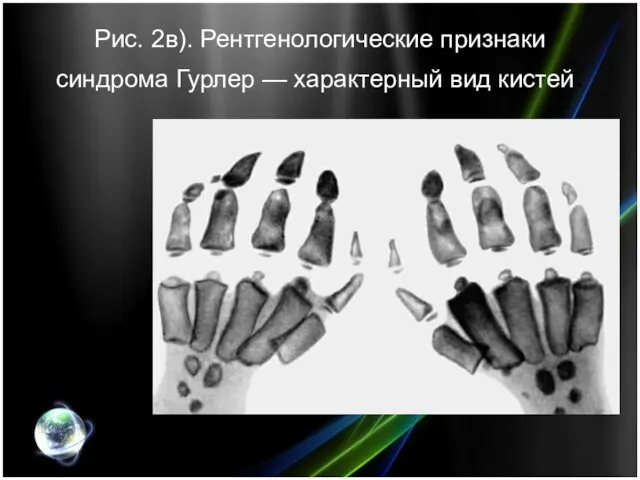
- 10. Рис. 2в). Рентгенологические признаки синдрома Гурлер — характерный вид кистей.
- 11. Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер). Впервые описан американским офтальмологом Шейе (Н.G. Scheie) в
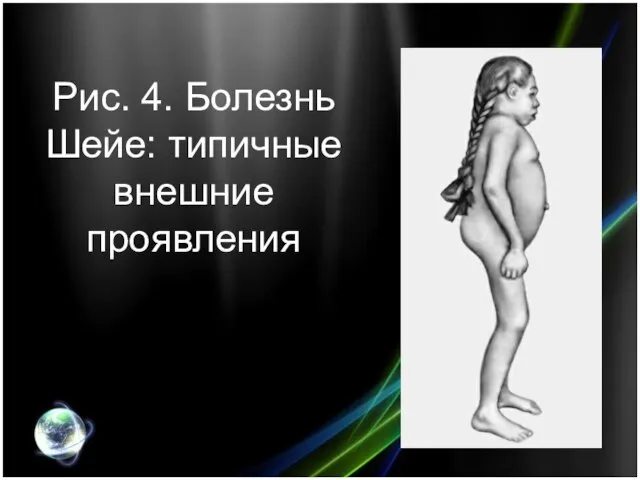
- 12. Рис. 4. Болезнь Шейе: типичные внешние проявления
- 13. Мукополисахаридоз типа II (синдром Гунтера). Клинические симптомы появляются позднее, чем при синдроме Гурлер (у детей старше
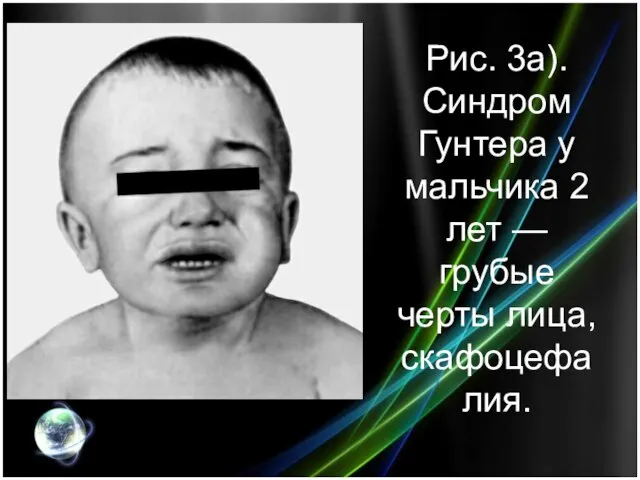
- 14. Рис. 3а). Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.
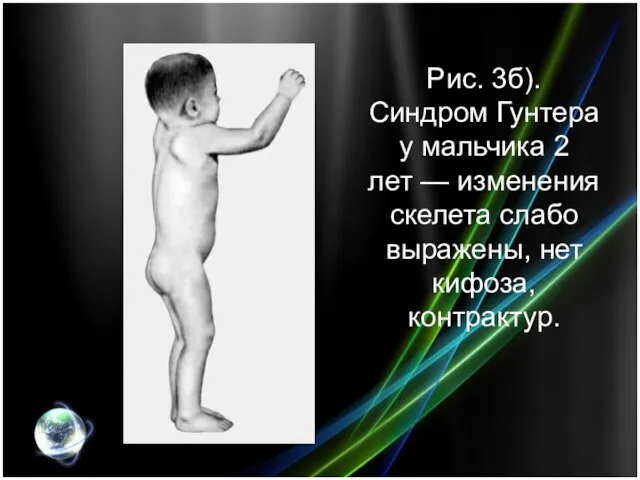
- 15. Рис. 3б). Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены, нет кифоза, контрактур.
- 16. Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо). Описан американским педиатром Санфилиппо (S.J. Sanfilippo) в 1963 г.
- 17. Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио). Заболевание в 1929 г. независимо друг от друга впервые
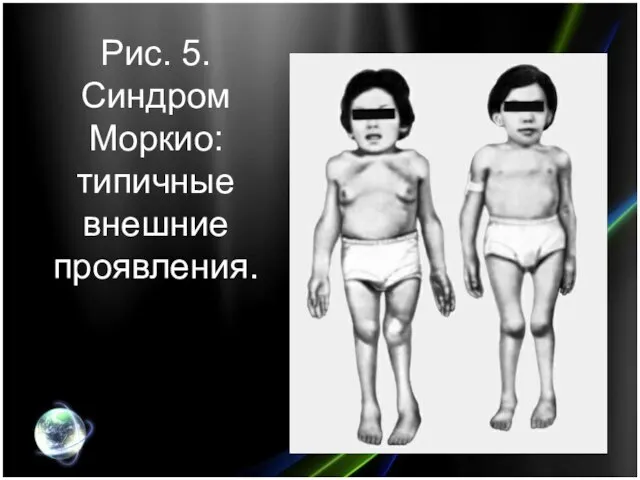
- 18. Рис. 5. Синдром Моркио: типичные внешние проявления.
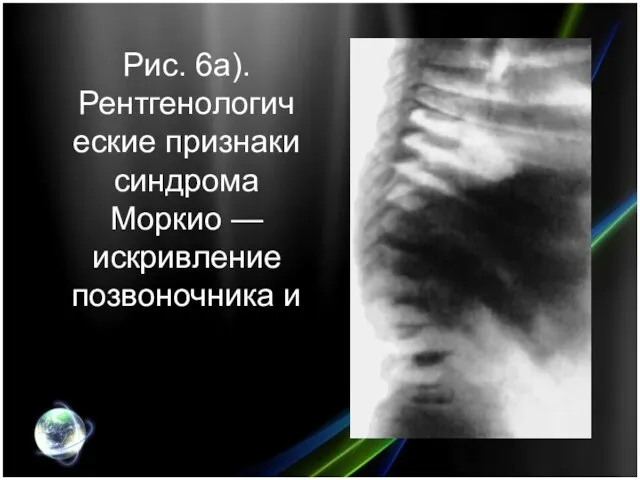
- 19. Рис. 6а). Рентгенологические признаки синдрома Моркио — искривление позвоночника и платиспондилия.
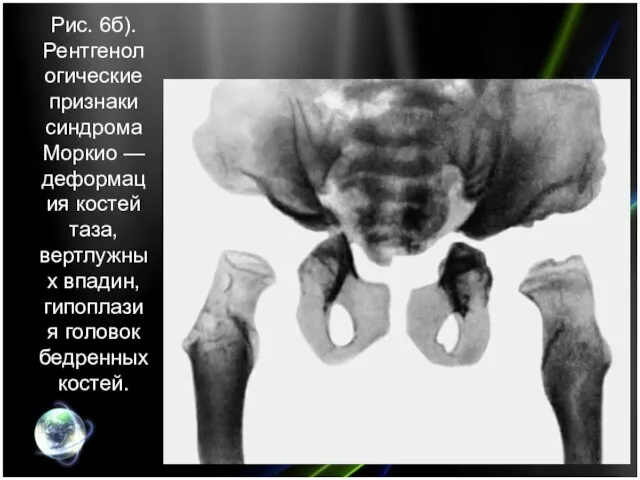
- 20. Рис. 6б). Рентгенологические признаки синдрома Моркио — деформация костей таза, вертлужных впадин, гипоплазия головок бедренных костей.
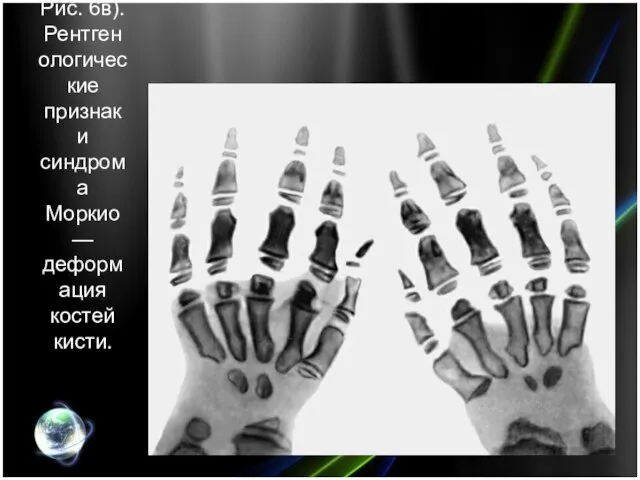
- 21. Рис. 6в). Рентгенологические признаки синдрома Моркио — деформация костей кисти.
- 22. Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами) в 1960 г. впервые описан
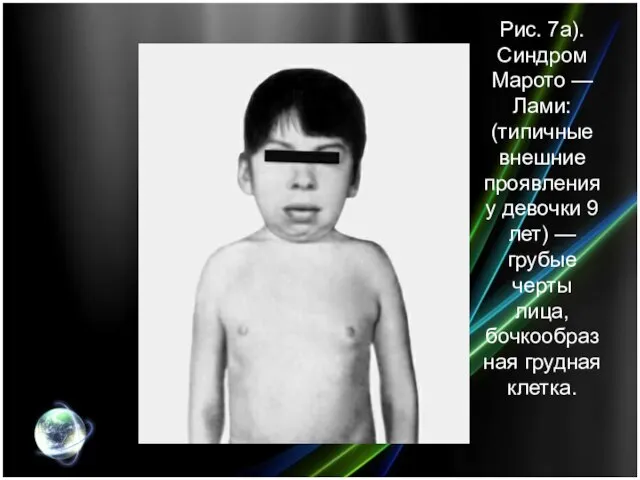
- 23. Рис. 7а). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — грубые черты
- 24. Рис. 7б). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — контрактуры верхних
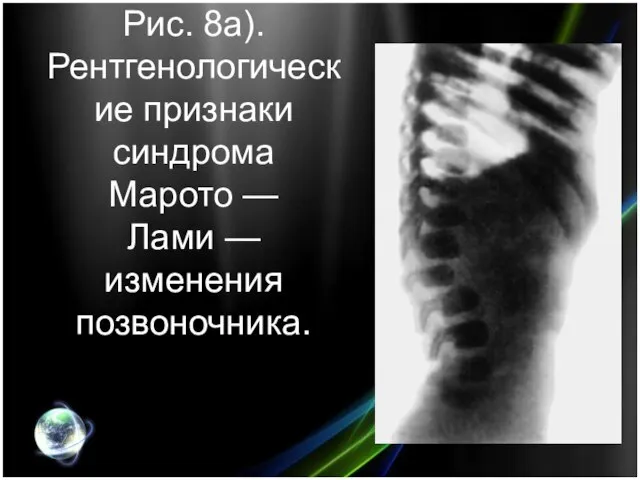
- 25. Рис. 8а). Рентгенологические признаки синдрома Марото — Лами — изменения позвоночника.
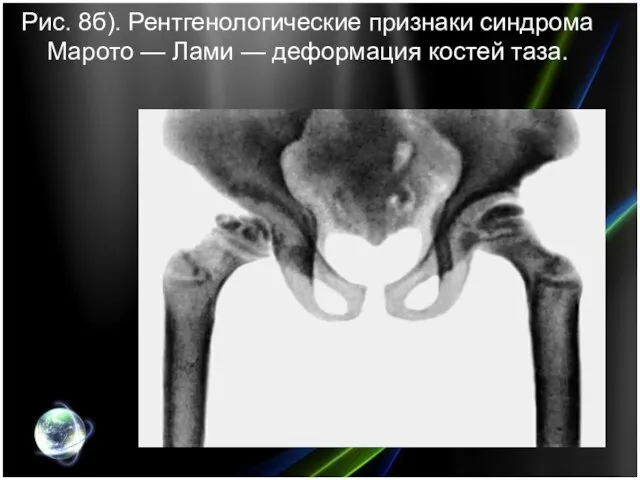
- 26. Рис. 8б). Рентгенологические признаки синдрома Марото — Лами — деформация костей таза.
- 27. Синдром Моркио-Улльриха. Генетически детерминированное заболевание - мукополисахаридоз - IV. Хондродистрофия характеризуется ослаблением связочного аппарата суставов, значительными
- 28. Редко встречаемые типы мукополисахаридозов Мукополисахаридоз типа VII (синдром Слая). Описан Слаем (W.S. Sly) в 1973 г.
- 29. Диагноз основывается на клинических проявлениях, данных рентгенологического исследования, определении экскреции с мочой гликозаминогликанов, исследовании активности специфических
- 31. Скачать презентацию
Слайд 2Мукополисахаридозы (мукополисахариды + -ōsis)
группа наследственных болезней соединительной ткани, обусловленных нарушением обмена гликозаминогликанов
Мукополисахаридозы (мукополисахариды + -ōsis)
группа наследственных болезней соединительной ткани, обусловленных нарушением обмена гликозаминогликанов

Слайд 3Мукополисахаридоз типа I-Н (синдром Гурлер).
Впервые описан немецким педиатром Гурлер (G. Hurler) в 1919 г.
Мукополисахаридоз типа I-Н (синдром Гурлер).
Впервые описан немецким педиатром Гурлер (G. Hurler) в 1919 г.

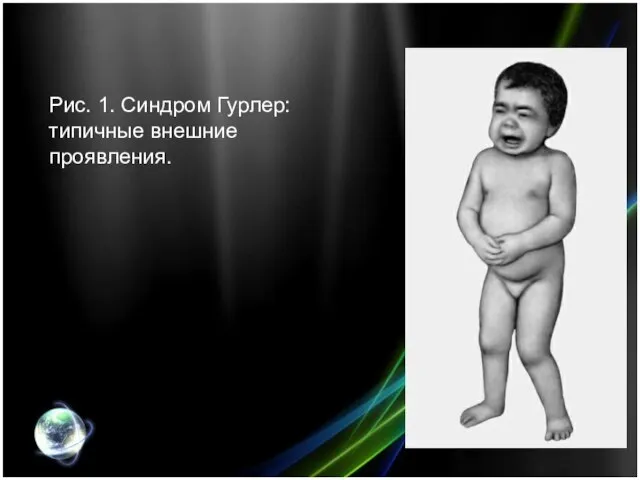
Слайд 4Рис. 1. Синдром Гурлер: типичные внешние проявления.
Рис. 1. Синдром Гурлер: типичные внешние проявления.
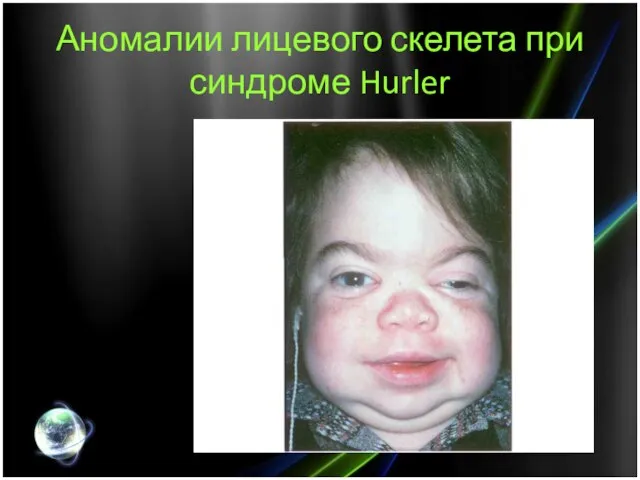
Слайд 5Аномалии лицевого скелета при синдроме Hurler
Аномалии лицевого скелета при синдроме Hurler
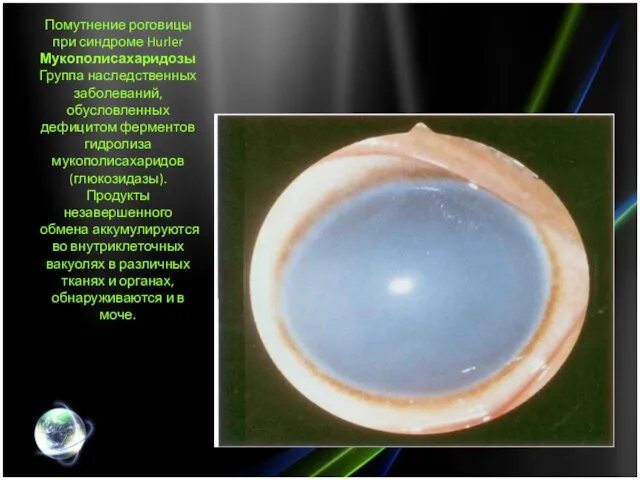
Слайд 6Помутнение роговицы при синдроме Hurler
Мукополисахаридозы
Группа наследственных заболеваний, обусловленных дефицитом ферментов гидролиза
Помутнение роговицы при синдроме Hurler Мукополисахаридозы Группа наследственных заболеваний, обусловленных дефицитом ферментов гидролиза
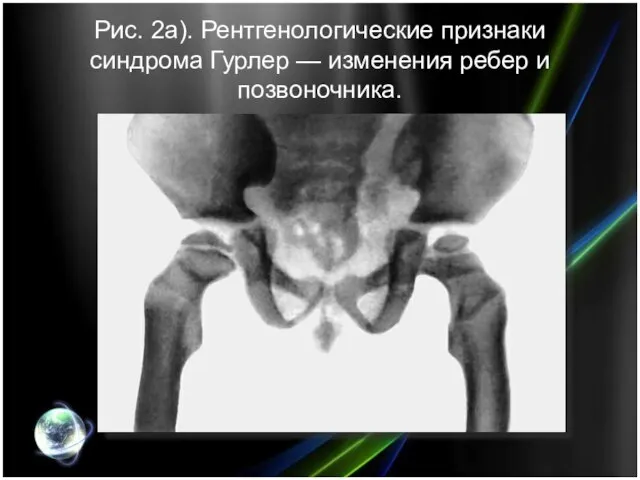
Слайд 7При рентгенологическом исследовании (рис. 2) выявляются характерные изменения позвонков и позвоночного столба
При рентгенологическом исследовании (рис. 2) выявляются характерные изменения позвонков и позвоночного столба

Слайд 8Рис. 2а). Рентгенологические признаки синдрома Гурлер — изменения ребер и позвоночника.
Рис. 2а). Рентгенологические признаки синдрома Гурлер — изменения ребер и позвоночника.
Слайд 9Рис. 2б). Рентгенологические признаки синдрома Гурлер — деформация таза и бедренных костей.
Рис. 2б). Рентгенологические признаки синдрома Гурлер — деформация таза и бедренных костей.
Слайд 10Рис. 2в). Рентгенологические признаки синдрома Гурлер — характерный вид кистей.
Рис. 2в). Рентгенологические признаки синдрома Гурлер — характерный вид кистей.
Слайд 11Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер).
Впервые описан американским офтальмологом Шейе
Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер).
Впервые описан американским офтальмологом Шейе

Слайд 12Рис. 4. Болезнь Шейе: типичные внешние проявления
Рис. 4. Болезнь Шейе: типичные внешние проявления
Слайд 13Мукополисахаридоз типа II (синдром Гунтера).
Клинические симптомы появляются позднее, чем при синдроме Гурлер
Мукополисахаридоз типа II (синдром Гунтера).
Клинические симптомы появляются позднее, чем при синдроме Гурлер

Слайд 14Рис. 3а). Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.
Рис. 3а). Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.
Слайд 15Рис. 3б). Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены,
Рис. 3б). Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены,
Слайд 16Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо).
Описан американским педиатром Санфилиппо (S.J. Sanfilippo) в
Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо).
Описан американским педиатром Санфилиппо (S.J. Sanfilippo) в

Слайд 17Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио).
Заболевание в 1929 г. независимо друг от
Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио).
Заболевание в 1929 г. независимо друг от

Слайд 18Рис. 5. Синдром Моркио: типичные внешние проявления.
Рис. 5. Синдром Моркио: типичные внешние проявления.
Слайд 19Рис. 6а). Рентгенологические признаки синдрома Моркио — искривление позвоночника и платиспондилия.
Рис. 6а). Рентгенологические признаки синдрома Моркио — искривление позвоночника и платиспондилия.
Слайд 20Рис. 6б). Рентгенологические признаки синдрома Моркио — деформация костей таза, вертлужных впадин, гипоплазия
Рис. 6б). Рентгенологические признаки синдрома Моркио — деформация костей таза, вертлужных впадин, гипоплазия
Слайд 21Рис. 6в). Рентгенологические признаки синдрома Моркио — деформация костей кисти.
Рис. 6в). Рентгенологические признаки синдрома Моркио — деформация костей кисти.
Слайд 22Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами)
в 1960 г. впервые описан
Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами)
в 1960 г. впервые описан

Слайд 23Рис. 7а). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) —
Рис. 7а). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) —
Слайд 24Рис. 7б). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) —
Рис. 7б). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) —

Слайд 25Рис. 8а). Рентгенологические признаки синдрома Марото — Лами — изменения позвоночника.
Рис. 8а). Рентгенологические признаки синдрома Марото — Лами — изменения позвоночника.
Слайд 26Рис. 8б). Рентгенологические признаки синдрома Марото — Лами — деформация костей таза.
Рис. 8б). Рентгенологические признаки синдрома Марото — Лами — деформация костей таза.
Слайд 27Синдром Моркио-Улльриха. Генетически детерминированное заболевание - мукополисахаридоз - IV. Хондродистрофия характеризуется ослаблением
Синдром Моркио-Улльриха. Генетически детерминированное заболевание - мукополисахаридоз - IV. Хондродистрофия характеризуется ослаблением

Слайд 28Редко встречаемые типы мукополисахаридозов
Мукополисахаридоз типа VII (синдром Слая). Описан Слаем (W.S. Sly) в
Редко встречаемые типы мукополисахаридозов
Мукополисахаридоз типа VII (синдром Слая). Описан Слаем (W.S. Sly) в

Слайд 29Диагноз основывается на клинических проявлениях, данных рентгенологического исследования, определении экскреции с мочой
Диагноз основывается на клинических проявлениях, данных рентгенологического исследования, определении экскреции с мочой

 Приверженность АРВТ Приверженность АРВТ
Приверженность АРВТ Приверженность АРВТ Противоопухолевая активность грибов
Противоопухолевая активность грибов Клинико-электркардиографическая признаки нарушений сердечного ритма, гипертрофии миокарда и желудочков, инфаркта миокарда
Клинико-электркардиографическая признаки нарушений сердечного ритма, гипертрофии миокарда и желудочков, инфаркта миокарда Лекарственные препараты, влияющие на систему гемостаза
Лекарственные препараты, влияющие на систему гемостаза Факторы риска неинфекционных заболеваний, увеличивающие вероятность ухудшения здоровья подростков и молодежи
Факторы риска неинфекционных заболеваний, увеличивающие вероятность ухудшения здоровья подростков и молодежи Медицинская реабилитация при острых и хронических заболеваниях лёгких
Медицинская реабилитация при острых и хронических заболеваниях лёгких Аппаратный педикюр. Школа Педикюра
Аппаратный педикюр. Школа Педикюра Делегированным синдромом Мюнхгаузена
Делегированным синдромом Мюнхгаузена Формирование таза и факторы вызывающие его деформацию
Формирование таза и факторы вызывающие его деформацию Обмен холестерола
Обмен холестерола Ша ле-фудс - программа детоксикации организма
Ша ле-фудс - программа детоксикации организма Профилактика тромбозов вен нижних конечностей
Профилактика тромбозов вен нижних конечностей Хирургиялық стоматологиялық қабылдауда болған науқасты тексеру әдістері
Хирургиялық стоматологиялық қабылдауда болған науқасты тексеру әдістері Гипотония (артериальная гипотензия)
Гипотония (артериальная гипотензия) Лечебно-диагностический процесс и сестринская деятельность при хроническом гепатите и циррозе печени
Лечебно-диагностический процесс и сестринская деятельность при хроническом гепатите и циррозе печени Профилактика контактной и имплантационной инфекции
Профилактика контактной и имплантационной инфекции Бруцеллез (болезнь Банга, мальтийская, средиземноморская или гибралтарская лихорадка)
Бруцеллез (болезнь Банга, мальтийская, средиземноморская или гибралтарская лихорадка) Гиперпролактинемия. Пролактин
Гиперпролактинемия. Пролактин Обезболивающие средства (анальгетики)
Обезболивающие средства (анальгетики) Апоплексия яичника
Апоплексия яичника Гигиена питания и воды
Гигиена питания и воды Травма живота
Травма живота Prenatal diagnosis
Prenatal diagnosis Рубцы кожи и их дерматокосметологическая коррекция
Рубцы кожи и их дерматокосметологическая коррекция Экспериментальная хирургия сердца
Экспериментальная хирургия сердца Шкала Сильвермана
Шкала Сильвермана Антифосфолипидный синдром
Антифосфолипидный синдром Питание здорового ребёнка
Питание здорового ребёнка