- Нефронофтиз и нефронофтиз-ассоциированные заболевания в детском возрасте

Содержание
- 2. АНАТОМИЯ ПОЧЕК Почки (лат. renes) – парный орган, поддерживающий постоянство внутренней среды организма путем мочеобразования. Они
- 3. СТРОЕНИЕ НЕФРОНА Основной морфологической и функциональной единицей строения почки является нефрон, состоящий из сосудистого клубочка и
- 4. Q61.5 НЕФРОНОФТИЗ Нефронофтиз (НФ), аутосомно-рецессивная медуллярная кистозная болезнь почек, является основной генетической причиной терминальной хронической почечной
- 5. ЭПИДЕМИОЛОГИЯ НФ Нефронофтиз (НФ) занимает лидирующее место в структуре наследственных причин хронической почечной недостаточности (ХПН) в
- 6. ЭТИОЛОГИЯ НФ Развитие НФ ассоциированно с мутацией генов нефроцистинов, инверсина. Мутации обуславливают нарушение функционирования цилий и
- 7. МОРФОЛОГИЧЕСКИЙ СУБСТРАТ НЕФРОНОФТИЗА – МУТАЦИИ ГЕНОВ МОНОЦИЛИЙ (ЦИЛИОПАТИИ) Цилии распространяются от поверхности клетки в экстрацелюлярное пространство
- 8. КЛАССИФИКАЦИЯ НФ В зависимости от возраста формирования ХПН (ХБП): Ювенильный Инфантильный Взрослый В зависимости от локализации
- 9. ПАТОГЕНЕЗ НЕФРОНОФТИЗА НФ – генетически гетерогенная группа болезней, характеризующаяся как изолированным поражением почек, так и полиорганным
- 10. ПАТОГЕНЕЗ НФ И НФ-АССОЦИИРОВАННЫХ СИНДРОМОВ Показано, что мутации NPHP5 (Сеньора–Локена синдром) и NPHP6 генов, экспрессирующихся на
- 11. НЕФРОНОФТИЗ 1‑ГО ТИПА (НЕФРОНОФТИЗ ФАНКОНИ) Нефронофтиз 1‑го типа — ювенильный, при котором мутантный ген NPHP1 локализован
- 12. НЕФРОНОФТИЗ 2‑ГО ТИПА Нефронофтиз 2‑го типа — инфантильный, обусловлен мутациями гена INV/NPHP2, локализованного на длинном плече
- 13. НЕФРОНОФТИЗ 3‑ГО ТИПА Нефронофтиз 3‑го типа — ювенильный, для него характерна мутация гена NPHP3, который картирован
- 14. НЕФРОНОФТИЗ 4‑ГО ТИПА Нефронофтиз 4‑го типа — взрослый, обусловлен мутациями гена NPHP4, расположенного на коротком плече
- 15. НФ-АССОЦИИРОВАННЫЕ СИНДРОМЫ Синдром Альстрема – наследственное заболевание, обусловленное мутацией гена ALMS1 (2p13.1) Частота встречаемости патологии не
- 16. НФ-АССОЦИИРОВАННЫЕ СИНДРОМЫ Синдром Барде-Бидля – аутосомно-рецессивное заболевание с вариабельной пенетрантностью, возникающее при наличии у ребенка одной
- 17. Бойхиса синдром (Сеньора–Бойхиса синдром) – наследственное заболевание, обусловленное мутацией гена TMEM67 (8q22.1) Частота встречаемости патологии не
- 18. Кранио-эктодермальная дисплазия – наследственное заболевание, обусловленное одной из мутаций генов: IFT122 (3q21.3-q22.1), IFT43 (14q24.3), WDR19 (4p14),
- 19. Элис-ван–Кревельда синдром (хондро-эктодермальная дисплазия) – наследственное заболевание, обусловленное одной из мутаций генов: EVC1 (4p16), EVC2 (4p16.2)
- 20. Джуберта синдром – наследственное заболевание, обусловленное одной из мутаций генов: AHI1 (6q23), NPHP1 (2q13), CEP290 (12q21),
- 21. Меккеля синдром - наследственное заболевание, обусловленное одной из мутаций генов: AHI1 (6q23), NPHP1 (2q13), CEP290 (12q21),
- 22. Джуберта синдром с окулоренальным дефектом (Арима синдром) – наследственное заболевание, обусловленное одной из мутаций генов: СС2D2A
- 23. Пигментный ретинит-гипопитуитаризм-нефронофтиз-костная дисплазия синдром (RHYNS) – наследственное заболевание неизвестной этиологии ( мутация не определена) Частота встречаемости
- 24. Сальдино–Майнцера синдром – наследственное заболевание, обусловленное одной из мутаций генов: IFT140 (16p13.3), IFT172 (2p23.3) Частота встречаемости
- 25. Сеньора–Локена синдром – наследственное заболевание, обусловленное одной из мутаций генов: NPHP1 (2q13), INVS (9q31.1), NPHP3 (3q21-22),
- 26. ДИАГНОСТИКА НЕФРОНОФТИЗА И НФ-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ Для постановки диагноза «нефронофтиз» необходимы: детальный анализ родословной пациента анализ клинических
- 27. ЛЕЧЕНИЕ НЕФРОНОФТИЗА В настоящее время не разработаны методы специфической терапии НФ Лечение является симптоматическим в период
- 28. Десмопрессин-резистентное нарушение концентрационной функции почек как первый клинический симптом НФ с последующим выявлением рецепторов к вазопрессину
- 29. ВЫВОДЫ Таким образом, НФ – генетически гетерогенная аутосомно-рецессивная болезнь почек, связанная с повреждением структуры и функции
- 31. ЛИТЕРАТУРА М.Е. Аксенова. Нефронофтиз и нефронофтиз-ассоциированные синдромы. Педиатрия; 2015; Т.94; № 3. М.С. Игнатова. Значение цилиопатий
- 33. Скачать презентацию
Слайд 2АНАТОМИЯ ПОЧЕК
Почки (лат. renes) – парный орган, поддерживающий постоянство внутренней среды организма путем
АНАТОМИЯ ПОЧЕК
Почки (лат. renes) – парный орган, поддерживающий постоянство внутренней среды организма путем

Слайд 3СТРОЕНИЕ НЕФРОНА
Основной морфологической и функциональной единицей строения почки является нефрон, состоящий из
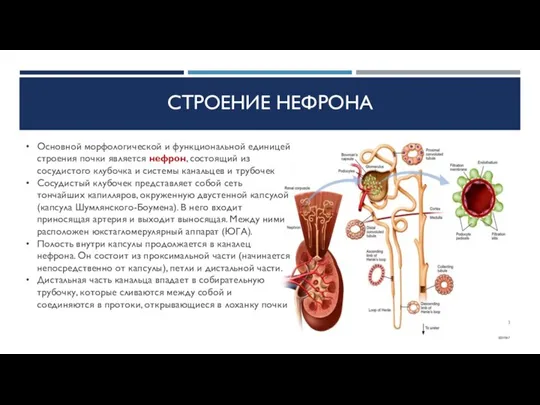
СТРОЕНИЕ НЕФРОНА
Основной морфологической и функциональной единицей строения почки является нефрон, состоящий из
Слайд 4Q61.5 НЕФРОНОФТИЗ
Нефронофтиз (НФ), аутосомно-рецессивная медуллярная кистозная болезнь почек, является основной генетической причиной
Q61.5 НЕФРОНОФТИЗ
Нефронофтиз (НФ), аутосомно-рецессивная медуллярная кистозная болезнь почек, является основной генетической причиной

Слайд 5ЭПИДЕМИОЛОГИЯ НФ
Нефронофтиз (НФ) занимает лидирующее место в структуре наследственных причин хронической почечной
ЭПИДЕМИОЛОГИЯ НФ
Нефронофтиз (НФ) занимает лидирующее место в структуре наследственных причин хронической почечной

Слайд 6ЭТИОЛОГИЯ НФ
Развитие НФ ассоциированно с мутацией генов нефроцистинов, инверсина. Мутации обуславливают нарушение
ЭТИОЛОГИЯ НФ
Развитие НФ ассоциированно с мутацией генов нефроцистинов, инверсина. Мутации обуславливают нарушение

Слайд 7МОРФОЛОГИЧЕСКИЙ СУБСТРАТ НЕФРОНОФТИЗА – МУТАЦИИ ГЕНОВ МОНОЦИЛИЙ (ЦИЛИОПАТИИ)
Цилии распространяются от поверхности клетки
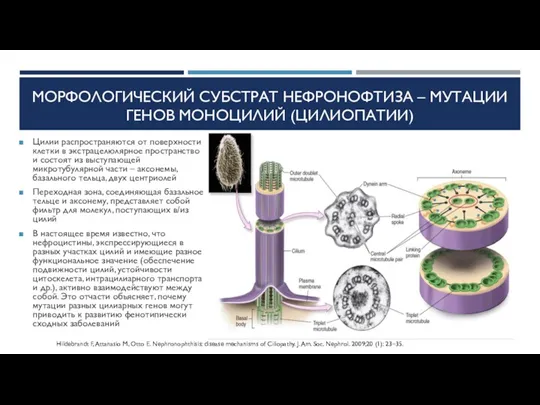
МОРФОЛОГИЧЕСКИЙ СУБСТРАТ НЕФРОНОФТИЗА – МУТАЦИИ ГЕНОВ МОНОЦИЛИЙ (ЦИЛИОПАТИИ)
Цилии распространяются от поверхности клетки
Слайд 8КЛАССИФИКАЦИЯ НФ
В зависимости от возраста формирования ХПН (ХБП):
Ювенильный
Инфантильный
Взрослый
В зависимости от локализации
КЛАССИФИКАЦИЯ НФ
В зависимости от возраста формирования ХПН (ХБП):
Ювенильный
Инфантильный
Взрослый
В зависимости от локализации
Слайд 9ПАТОГЕНЕЗ НЕФРОНОФТИЗА
НФ – генетически гетерогенная группа болезней, характеризующаяся как изолированным поражением почек,
ПАТОГЕНЕЗ НЕФРОНОФТИЗА
НФ – генетически гетерогенная группа болезней, характеризующаяся как изолированным поражением почек,

Слайд 10ПАТОГЕНЕЗ НФ И НФ-АССОЦИИРОВАННЫХ СИНДРОМОВ
Показано, что мутации NPHP5 (Сеньора–Локена синдром) и NPHP6
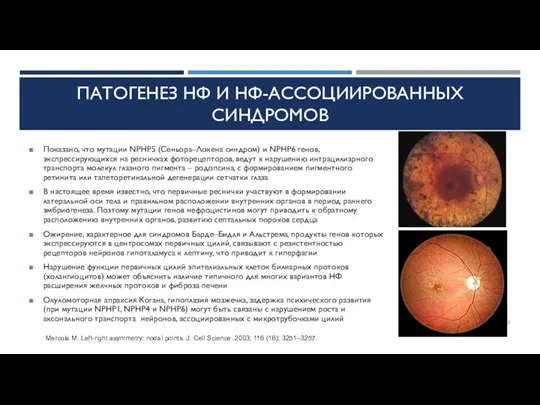
ПАТОГЕНЕЗ НФ И НФ-АССОЦИИРОВАННЫХ СИНДРОМОВ
Показано, что мутации NPHP5 (Сеньора–Локена синдром) и NPHP6
Слайд 11НЕФРОНОФТИЗ 1‑ГО ТИПА (НЕФРОНОФТИЗ ФАНКОНИ)
Нефронофтиз 1‑го типа — ювенильный, при котором мутантный
НЕФРОНОФТИЗ 1‑ГО ТИПА (НЕФРОНОФТИЗ ФАНКОНИ)
Нефронофтиз 1‑го типа — ювенильный, при котором мутантный

Слайд 12НЕФРОНОФТИЗ 2‑ГО ТИПА
Нефронофтиз 2‑го типа — инфантильный, обусловлен мутациями гена INV/NPHP2, локализованного
НЕФРОНОФТИЗ 2‑ГО ТИПА
Нефронофтиз 2‑го типа — инфантильный, обусловлен мутациями гена INV/NPHP2, локализованного

Слайд 13НЕФРОНОФТИЗ 3‑ГО ТИПА
Нефронофтиз 3‑го типа — ювенильный, для него характерна мутация гена
НЕФРОНОФТИЗ 3‑ГО ТИПА
Нефронофтиз 3‑го типа — ювенильный, для него характерна мутация гена

Слайд 14НЕФРОНОФТИЗ 4‑ГО ТИПА
Нефронофтиз 4‑го типа — взрослый, обусловлен мутациями гена NPHP4, расположенного
НЕФРОНОФТИЗ 4‑ГО ТИПА
Нефронофтиз 4‑го типа — взрослый, обусловлен мутациями гена NPHP4, расположенного

Слайд 15НФ-АССОЦИИРОВАННЫЕ СИНДРОМЫ
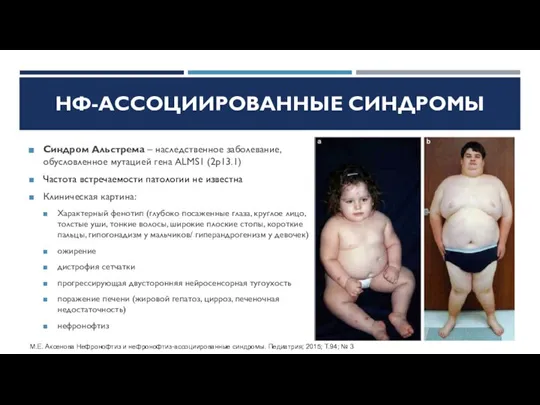
Синдром Альстрема – наследственное заболевание, обусловленное мутацией гена ALMS1 (2p13.1)
Частота
НФ-АССОЦИИРОВАННЫЕ СИНДРОМЫ
Синдром Альстрема – наследственное заболевание, обусловленное мутацией гена ALMS1 (2p13.1)
Частота
Слайд 16НФ-АССОЦИИРОВАННЫЕ СИНДРОМЫ
Синдром Барде-Бидля – аутосомно-рецессивное заболевание с вариабельной пенетрантностью, возникающее при наличии
НФ-АССОЦИИРОВАННЫЕ СИНДРОМЫ
Синдром Барде-Бидля – аутосомно-рецессивное заболевание с вариабельной пенетрантностью, возникающее при наличии

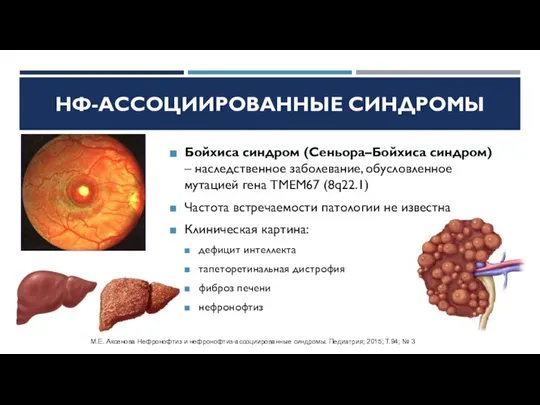
Слайд 17Бойхиса синдром (Сеньора–Бойхиса синдром) – наследственное заболевание, обусловленное мутацией гена TMEM67 (8q22.1)
Частота
Бойхиса синдром (Сеньора–Бойхиса синдром) – наследственное заболевание, обусловленное мутацией гена TMEM67 (8q22.1)
Частота
Слайд 18Кранио-эктодермальная дисплазия – наследственное заболевание, обусловленное одной из мутаций генов: IFT122 (3q21.3-q22.1),
Кранио-эктодермальная дисплазия – наследственное заболевание, обусловленное одной из мутаций генов: IFT122 (3q21.3-q22.1),

Слайд 19Элис-ван–Кревельда синдром (хондро-эктодермальная дисплазия) – наследственное заболевание, обусловленное одной из мутаций генов:
Элис-ван–Кревельда синдром (хондро-эктодермальная дисплазия) – наследственное заболевание, обусловленное одной из мутаций генов:

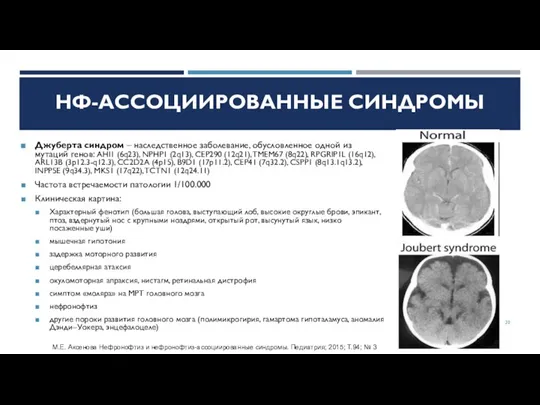
Слайд 20Джуберта синдром – наследственное заболевание, обусловленное одной из мутаций генов: AHI1 (6q23),
Джуберта синдром – наследственное заболевание, обусловленное одной из мутаций генов: AHI1 (6q23),
Слайд 21Меккеля синдром - наследственное заболевание, обусловленное одной из мутаций генов: AHI1 (6q23),
Меккеля синдром - наследственное заболевание, обусловленное одной из мутаций генов: AHI1 (6q23),

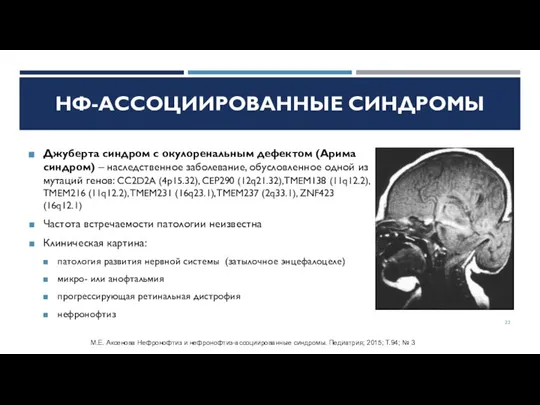
Слайд 22Джуберта синдром с окулоренальным дефектом (Арима синдром) – наследственное заболевание, обусловленное одной
Джуберта синдром с окулоренальным дефектом (Арима синдром) – наследственное заболевание, обусловленное одной
Слайд 23Пигментный ретинит-гипопитуитаризм-нефронофтиз-костная дисплазия синдром (RHYNS) – наследственное заболевание неизвестной этиологии ( мутация
Пигментный ретинит-гипопитуитаризм-нефронофтиз-костная дисплазия синдром (RHYNS) – наследственное заболевание неизвестной этиологии ( мутация

Слайд 24Сальдино–Майнцера синдром – наследственное заболевание, обусловленное одной из мутаций генов: IFT140 (16p13.3),
Сальдино–Майнцера синдром – наследственное заболевание, обусловленное одной из мутаций генов: IFT140 (16p13.3),
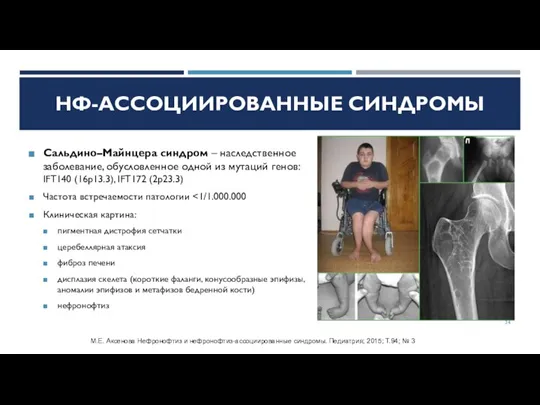
Слайд 25Сеньора–Локена синдром – наследственное заболевание, обусловленное одной из мутаций генов: NPHP1 (2q13),
Сеньора–Локена синдром – наследственное заболевание, обусловленное одной из мутаций генов: NPHP1 (2q13),

Слайд 26ДИАГНОСТИКА НЕФРОНОФТИЗА И НФ-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ
Для постановки диагноза «нефронофтиз» необходимы:
детальный анализ родословной
ДИАГНОСТИКА НЕФРОНОФТИЗА И НФ-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ
Для постановки диагноза «нефронофтиз» необходимы:
детальный анализ родословной

Слайд 27ЛЕЧЕНИЕ НЕФРОНОФТИЗА
В настоящее время не разработаны методы специфической терапии НФ
Лечение является
ЛЕЧЕНИЕ НЕФРОНОФТИЗА
В настоящее время не разработаны методы специфической терапии НФ
Лечение является
Слайд 28Десмопрессин-резистентное нарушение концентрационной функции почек как первый клинический симптом НФ с последующим
Десмопрессин-резистентное нарушение концентрационной функции почек как первый клинический симптом НФ с последующим

Слайд 29ВЫВОДЫ
Таким образом, НФ – генетически гетерогенная аутосомно-рецессивная болезнь почек, связанная с повреждением
ВЫВОДЫ
Таким образом, НФ – генетически гетерогенная аутосомно-рецессивная болезнь почек, связанная с повреждением


Слайд 31ЛИТЕРАТУРА
М.Е. Аксенова. Нефронофтиз и нефронофтиз-ассоциированные синдромы. Педиатрия; 2015; Т.94; № 3.
М.С. Игнатова.
ЛИТЕРАТУРА
М.Е. Аксенова. Нефронофтиз и нефронофтиз-ассоциированные синдромы. Педиатрия; 2015; Т.94; № 3.
М.С. Игнатова.

 Профилактика вирусных инфекций у детей
Профилактика вирусных инфекций у детей Значение эндокринной системы
Значение эндокринной системы Понятие болезни
Понятие болезни Невынашивание беременности. Преждевременные роды. Перенашивание беременности
Невынашивание беременности. Преждевременные роды. Перенашивание беременности Рациональное питание
Рациональное питание Особливості акушерської та перинатальної патології на тлі загрози передчасних пологів
Особливості акушерської та перинатальної патології на тлі загрози передчасних пологів Аутоиммунный гепатит
Аутоиммунный гепатит Неинфекционные заболевания
Неинфекционные заболевания Иммунитет
Иммунитет Профилактический массаж
Профилактический массаж Клинический случай. Операция Бенталла - Де Боно
Клинический случай. Операция Бенталла - Де Боно Несовершенный остеогенез
Несовершенный остеогенез Редкие формы внематочной беременности: абдоминальная и шеечная
Редкие формы внематочной беременности: абдоминальная и шеечная Острая кишечная непроходимость
Острая кишечная непроходимость Микозы человека
Микозы человека Понятие гиподинамии, гипердинамии
Понятие гиподинамии, гипердинамии Инфаркт миокарда. Медицинские аспекты. Модуль 2
Инфаркт миокарда. Медицинские аспекты. Модуль 2 Пути восстановления ВПФ
Пути восстановления ВПФ ВИЧ, или вирус иммунодефицита человека
ВИЧ, или вирус иммунодефицита человека Строение сердечно-сосудистой системы. (Тема 2.3)
Строение сердечно-сосудистой системы. (Тема 2.3) Некоторые проблемы и решения на примере ВИЧ-инфекции и алкоголя
Некоторые проблемы и решения на примере ВИЧ-инфекции и алкоголя Патофизиология нарушений сознания. Угнетение сознания. Кома. Занятие 11
Патофизиология нарушений сознания. Угнетение сознания. Кома. Занятие 11 Құпия сөзбен қорғау
Құпия сөзбен қорғау Патофизиология седины
Патофизиология седины Затруднение носового дыхания
Затруднение носового дыхания Основы хирургии. Раны и их классификация. Первая помощь. Десмургия (наука о повязках)
Основы хирургии. Раны и их классификация. Первая помощь. Десмургия (наука о повязках) Поверхностный кариес
Поверхностный кариес Первая помощь при обморожении
Первая помощь при обморожении