- Screening of possible antiviral peptides to bind SARS Covid 19 spike protein

Содержание
- 2. Content Intoduction Sructure of SARS-CoV-2 Aim of the study Methods VMD CABS Dock 5. Results
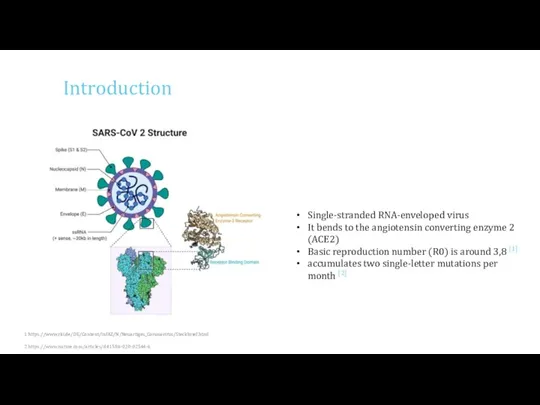
- 3. Introduction 1 https://www.rki.de/DE/Content/InfAZ/N/Neuartiges_Coronavirus/Steckbrief.html 2 https://www.nature.com/articles/d41586-020-02544-6 Single-stranded RNA-enveloped virus It bends to the angiotensin converting enzyme 2
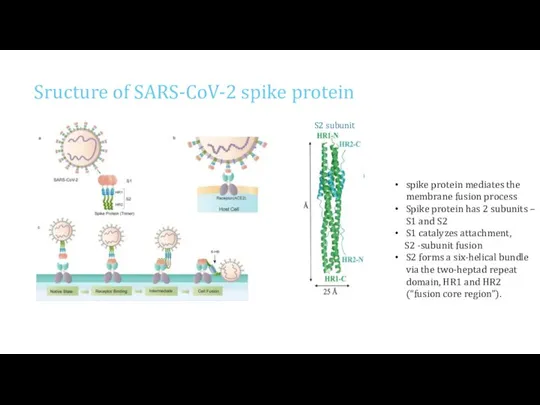
- 4. Sructure of SARS-CoV-2 spike protein spike protein mediates the membrane fusion process Spike protein has 2
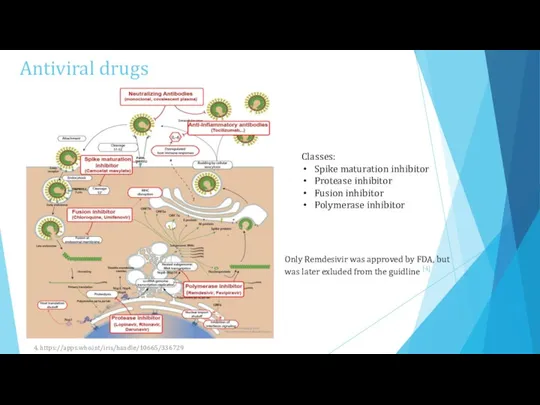
- 5. Antiviral drugs Classes: Spike maturation inhibitor Protease inhibitor Fusion inhibitor Polymerase inhibitor Only Remdesivir was approved
- 7. Aim of the study to screen a list of peptides which were designed to bind the
- 8. Methods
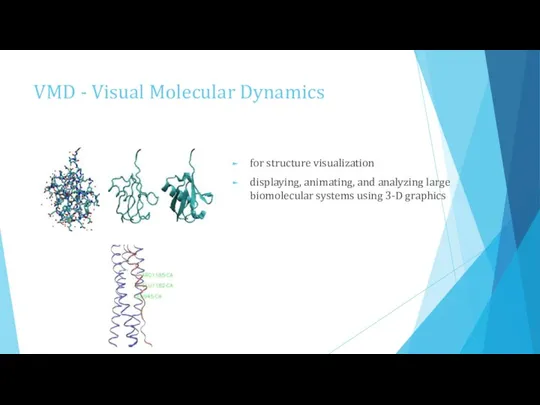
- 9. VMD - Visual Molecular Dynamics for structure visualization displaying, animating, and analyzing large biomolecular systems using
- 10. CABS Dock for protein-peptide docking coarse-grained model (it decreases a time of long simulations) advantages: Can
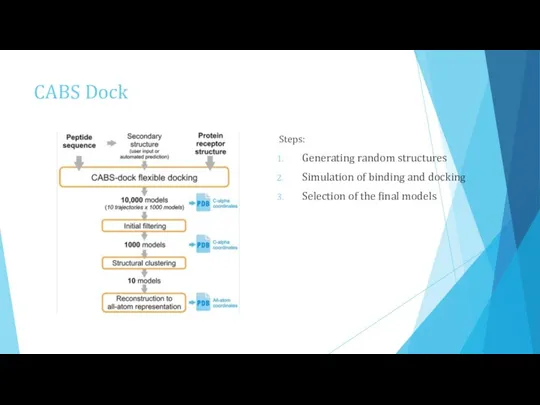
- 11. CABS Dock Steps: Generating random structures Simulation of binding and docking Selection of the final models
- 12. Results Original structure: Fraction of contacts – 51 % (cutoff – 5Å) number of residues in
- 13. Original Model 4 200 cycles were also chosen for docking the derivatives.
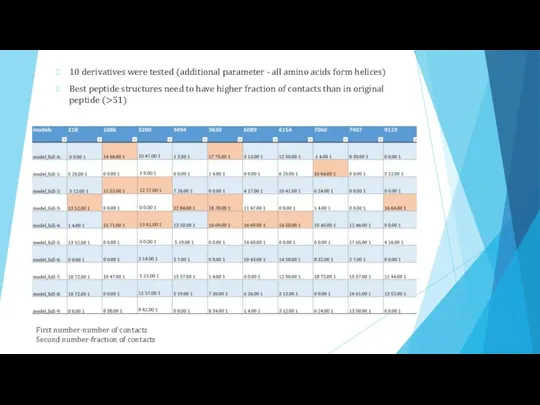
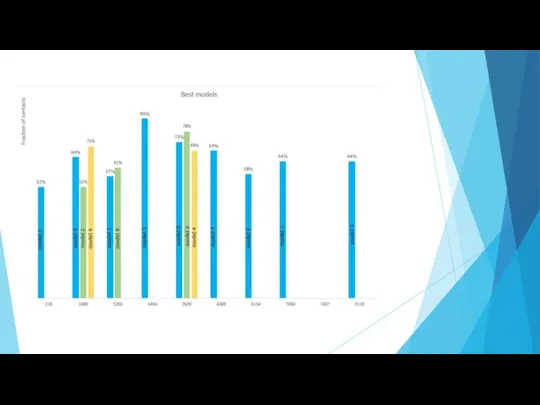
- 14. 10 derivatives were tested (additional parameter - all amino acids form helices) Best peptide structures need
- 16. Ranking according to the predicted number of models, fractions and position of a modal in a
- 17. PPI-Affinity The most promising candidates: DERIVATIVE_1086 DERIVATIVE_3200 DERIVATIVE_3630 original -8.4
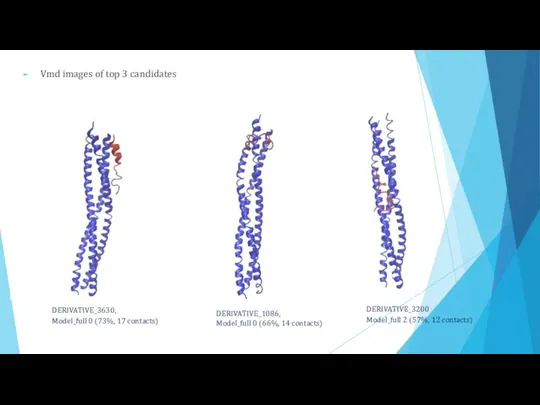
- 18. Vmd images of top 3 candidates DERIVATIVE_3630, Model_full 0 (73%, 17 contacts) DERIVATIVE_1086, Model_full 0 (66%,
- 20. Скачать презентацию

Слайд 3Introduction
1 https://www.rki.de/DE/Content/InfAZ/N/Neuartiges_Coronavirus/Steckbrief.html
2 https://www.nature.com/articles/d41586-020-02544-6
Single-stranded RNA-enveloped virus
It bends to the angiotensin converting enzyme
Introduction
1 https://www.rki.de/DE/Content/InfAZ/N/Neuartiges_Coronavirus/Steckbrief.html
2 https://www.nature.com/articles/d41586-020-02544-6
Single-stranded RNA-enveloped virus
It bends to the angiotensin converting enzyme
Слайд 4Sructure of SARS-CoV-2 spike protein
spike protein mediates the membrane fusion process
Spike protein
Sructure of SARS-CoV-2 spike protein
spike protein mediates the membrane fusion process
Spike protein
Слайд 5Antiviral drugs
Classes:
Spike maturation inhibitor
Protease inhibitor
Fusion inhibitor
Polymerase inhibitor
Only Remdesivir was approved by FDA,
Antiviral drugs
Classes:
Spike maturation inhibitor
Protease inhibitor
Fusion inhibitor
Polymerase inhibitor
Only Remdesivir was approved by FDA,
Слайд 7Aim of the study
to screen a list of peptides which were designed
Aim of the study
to screen a list of peptides which were designed

Слайд 8Methods
Methods

Слайд 9VMD - Visual Molecular Dynamics
for structure visualization
displaying, animating, and analyzing large biomolecular
VMD - Visual Molecular Dynamics
for structure visualization
displaying, animating, and analyzing large biomolecular
Слайд 10CABS Dock
for protein-peptide docking
coarse-grained model (it decreases a time of long simulations)
advantages:
Can
CABS Dock
for protein-peptide docking
coarse-grained model (it decreases a time of long simulations)
advantages:
Can

Слайд 11CABS Dock
Steps:
Generating random structures
Simulation of binding and docking
Selection of the final
CABS Dock
Steps:
Generating random structures
Simulation of binding and docking
Selection of the final
Слайд 12Results
Original structure:
Fraction of contacts – 51 % (cutoff – 5Å)
number of residues
Results
Original structure:
Fraction of contacts – 51 % (cutoff – 5Å)
number of residues

Слайд 13Original Model 4
200 cycles were also chosen for docking the derivatives.
Original Model 4
200 cycles were also chosen for docking the derivatives.

Слайд 1410 derivatives were tested (additional parameter - all amino acids form helices)
Best
10 derivatives were tested (additional parameter - all amino acids form helices)
Best
Слайд 16Ranking according to the predicted number of models, fractions and position of
Ranking according to the predicted number of models, fractions and position of

Слайд 17PPI-Affinity
The most promising candidates:
DERIVATIVE_1086
DERIVATIVE_3200
DERIVATIVE_3630
original
-8.4
PPI-Affinity
The most promising candidates:
DERIVATIVE_1086
DERIVATIVE_3200
DERIVATIVE_3630
original
-8.4

Слайд 18Vmd images of top 3 candidates
DERIVATIVE_3630,
Model_full 0 (73%, 17 contacts)
DERIVATIVE_1086,
Model_full
Vmd images of top 3 candidates
DERIVATIVE_3630,
Model_full 0 (73%, 17 contacts)
DERIVATIVE_1086,
Model_full
 Радиационное излучение. Заболевания под его влиянием
Радиационное излучение. Заболевания под его влиянием Мониторинг физического состояния
Мониторинг физического состояния Дифференцированный подход к назначению питания детям раннего возраста
Дифференцированный подход к назначению питания детям раннего возраста Пастерель. Збудник
Пастерель. Збудник Герпесвирусные инфекции V, VI, VII, VIII типов
Герпесвирусные инфекции V, VI, VII, VIII типов Плацентарная недостаточность
Плацентарная недостаточность Обезболивающие и сосудосуживающие средства
Обезболивающие и сосудосуживающие средства Трудный дыхательный путь. Методы его преодоления с позиции анестезиолога
Трудный дыхательный путь. Методы его преодоления с позиции анестезиолога Бақылаудағы науқастың анамнезін жинау және жергілікті статусын сипаттау
Бақылаудағы науқастың анамнезін жинау және жергілікті статусын сипаттау Пищеварение в кишечнике
Пищеварение в кишечнике Облитерирующий тромбангиит (болезнь Винивартера — Бюргера)
Облитерирующий тромбангиит (болезнь Винивартера — Бюргера) Антибиотики
Антибиотики Экстремальные состояния
Экстремальные состояния Аллергические реакции
Аллергические реакции Врач-стоматолог Профессия - дарить улыбку
Врач-стоматолог Профессия - дарить улыбку Типичный гемолитико-уремический синдром с тяжелым течением. Разбор клинического случая
Типичный гемолитико-уремический синдром с тяжелым течением. Разбор клинического случая Антиметаболиты. Пролиферация клеток и апоптоз
Антиметаболиты. Пролиферация клеток и апоптоз Есекжем
Есекжем Тонзилит(баспа)
Тонзилит(баспа) Клинические аспекты применения современных антидепрессантов в психиатрической практике
Клинические аспекты применения современных антидепрессантов в психиатрической практике Биопсия при проведении УЗИ
Биопсия при проведении УЗИ Правила личной гигиены
Правила личной гигиены Онтофилогенетические пороки развития скелета
Онтофилогенетические пороки развития скелета Дизайн лікарських засобів. Оптимізація ліду (частина 2)
Дизайн лікарських засобів. Оптимізація ліду (частина 2) Клиническая анатомия спинного мозга и конского хвоста. Основные синдромы поражения
Клиническая анатомия спинного мозга и конского хвоста. Основные синдромы поражения Опухоли мочеточника
Опухоли мочеточника Infection control. Final exam
Infection control. Final exam Определение локального статуса хирургического больного
Определение локального статуса хирургического больного