- Разбор задания. Часть i

Содержание
- 2. Общая схема решения поиск SNP аннотирование SNP Анализ результатов В каких позициях референс и наши данные
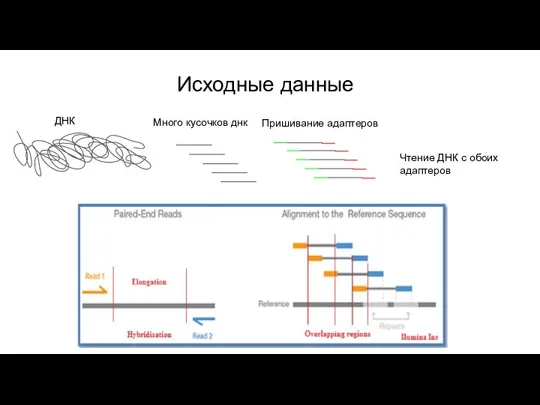
- 3. Исходные данные ДНК Много кусочков днк Пришивание адаптеров Чтение ДНК с обоих адаптеров
- 4. Исходные данные - подготовка Выбор ридов с X хромосомы Де-картирование @SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65 fastq
- 5. Инструменты для реализации общего решения можно подобрать совершенно разные!! UGENE Galaxy …. Bwa mem strelka openCravat
- 6. Картирование (bwa mem) #!/bin/bash while read p; do echo $p echo "#########################Started mapping#######################" bwa mem -R
- 7. Результат картирования (SAM/BAM) https://bioinformatics-core-shared-training.github.io/cruk-summer-school-2017/Day1/Session5-alignedReads.html
- 8. Поиск SNP (strelka) #!/bin/bash # configuration ./strelka-2.9.10.centos6_x86_64/bin/configureStrelkaGermlineWorkflow.py \ --bam ./sensitive_1.MD.bam \ --bam ./sensitive_2.MD.bam \ --bam ./sensitive_3.MD.bam
- 9. Результаты поиска SNP (VCF формат) #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT resist1 resist2
- 11. Скачать презентацию
Слайд 2Общая схема решения
поиск SNP
аннотирование SNP
Анализ результатов
В каких позициях референс и наши данные
Общая схема решения
поиск SNP
аннотирование SNP
Анализ результатов
В каких позициях референс и наши данные

Слайд 3Исходные данные
ДНК
Много кусочков днк
Пришивание адаптеров
Чтение ДНК с обоих адаптеров
Исходные данные
ДНК
Много кусочков днк
Пришивание адаптеров
Чтение ДНК с обоих адаптеров
Слайд 4Исходные данные - подготовка
Выбор ридов с X хромосомы
Де-картирование
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
fastq
Исходные данные - подготовка
Выбор ридов с X хромосомы
Де-картирование
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
fastq

Слайд 5Инструменты для реализации общего решения можно подобрать совершенно разные!!
UGENE
Galaxy
….
Bwa mem
strelka
openCravat
R
Инструменты для реализации общего решения можно подобрать совершенно разные!!
UGENE
Galaxy
….
Bwa mem
strelka
openCravat
R

Слайд 6Картирование (bwa mem)
#!/bin/bash
while read p; do
echo $p
echo "#########################Started mapping#######################"
bwa
Картирование (bwa mem)
#!/bin/bash
while read p; do
echo $p
echo "#########################Started mapping#######################"
bwa

Слайд 7Результат картирования (SAM/BAM)
https://bioinformatics-core-shared-training.github.io/cruk-summer-school-2017/Day1/Session5-alignedReads.html
Результат картирования (SAM/BAM)
https://bioinformatics-core-shared-training.github.io/cruk-summer-school-2017/Day1/Session5-alignedReads.html

Слайд 8Поиск SNP (strelka)
#!/bin/bash
# configuration
./strelka-2.9.10.centos6_x86_64/bin/configureStrelkaGermlineWorkflow.py \
--bam ./sensitive_1.MD.bam \
--bam ./sensitive_2.MD.bam \
--bam ./sensitive_3.MD.bam \
--bam ./sensitive_4.MD.bam
Поиск SNP (strelka)
#!/bin/bash
# configuration
./strelka-2.9.10.centos6_x86_64/bin/configureStrelkaGermlineWorkflow.py \
--bam ./sensitive_1.MD.bam \
--bam ./sensitive_2.MD.bam \
--bam ./sensitive_3.MD.bam \
--bam ./sensitive_4.MD.bam

Слайд 9Результаты поиска SNP (VCF формат)
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT resist1 resist2 resist3 resist4 resist5 resist6
chrX 19942 . G A 2 PASS SNVHPOL=2;MQ=50 GT:GQ:GQX:DP:DPF:AD:ADF:ADR:SB:FT:PL
0/1:29:4:4:0:3,1:3,1:0,0:0.0:PASS:32,0,75
6 колонок (по одной для каждого образца)
Результаты поиска SNP (VCF формат)
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT resist1 resist2 resist3 resist4 resist5 resist6
chrX 19942 . G A 2 PASS SNVHPOL=2;MQ=50 GT:GQ:GQX:DP:DPF:AD:ADF:ADR:SB:FT:PL
0/1:29:4:4:0:3,1:3,1:0,0:0.0:PASS:32,0,75
6 колонок (по одной для каждого образца)

 Урок технологии
Урок технологии Презентация на тему Гимнастика 5-11 класс
Презентация на тему Гимнастика 5-11 класс  Законы перспективы
Законы перспективы Домовой. 2 класс
Домовой. 2 класс Работа с бумагой. Нарцисс
Работа с бумагой. Нарцисс Воспитание ответственного отношения школьников к своему здоровью средствами физической культуры
Воспитание ответственного отношения школьников к своему здоровью средствами физической культуры Осенний лист. Фотокросс
Осенний лист. Фотокросс Определение понятия. Что такое понятие?
Определение понятия. Что такое понятие? Космическая викторина
Космическая викторина Книга сказок, рассказов и загадок про полицию. Темы: Если б я был полицейским…, Каким должен быть настоящий полицейский
Книга сказок, рассказов и загадок про полицию. Темы: Если б я был полицейским…, Каким должен быть настоящий полицейский Презентация на тему Расписание дня школьника
Презентация на тему Расписание дня школьника  Профессиональный модуль 03 Классное руководство
Профессиональный модуль 03 Классное руководство Понятие подшивание. Подшивание вручную прямыми стежками
Понятие подшивание. Подшивание вручную прямыми стежками Шаблон (пример) к очному этапу
Шаблон (пример) к очному этапу Методология научного исследования
Методология научного исследования Калядная замалёўка Ехала каляда ў чырвоным вазочку
Калядная замалёўка Ехала каляда ў чырвоным вазочку Разработчик программного обеспечения
Разработчик программного обеспечения Что может быть важнее учебы
Что может быть важнее учебы Как мы провели лето
Как мы провели лето Учимся рисовать. Заяц
Учимся рисовать. Заяц Родительское собрание в младших классах
Родительское собрание в младших классах Симметрия
Симметрия Дидактические требования к уроку
Дидактические требования к уроку Собрание MGRI ЕAGE Student Chapter
Собрание MGRI ЕAGE Student Chapter Типы и виды уроков
Типы и виды уроков Викторина: Экономика и мы
Викторина: Экономика и мы Жоғары метеп педагогикасының әдіснамасы
Жоғары метеп педагогикасының әдіснамасы Бумажный планер
Бумажный планер