- Наследственная недостаточность Альфа-1-антитрипсина

Содержание
- 3. Наследственная недостаточность α1антитрипсина (α1АТ) – генетическое заболевание, клинически проявляющееся эмфиземой легких, циррозом печени и значительно реже
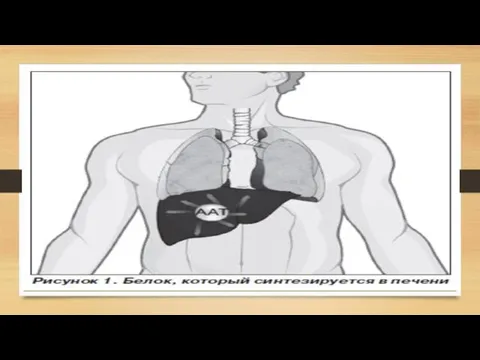
- 4. Общие сведения об альфа-1-антитриписине А1АТ представляет собой низкомолекулярный гликопротеин, состоящий из 394 аминокислотных остатков и трех
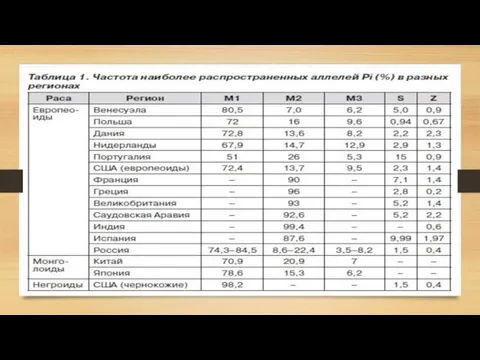
- 5. Генетический полиморфизм альфа-1-антитриписина За продукцию А1АТ отвечает ген, расположенный на хромосоме 14q32.1, называемый SERPINA1 (serpin peptidase
- 6. Есть несколько форм и степеней дефицита, которые главным образом зависят от того, сколько (1 или 2
- 8. Варианты наследования гена альфа-1-антитрипсина ММ — оба варианта гена здоровы MS или MZ (PiMS, PiMZ) —
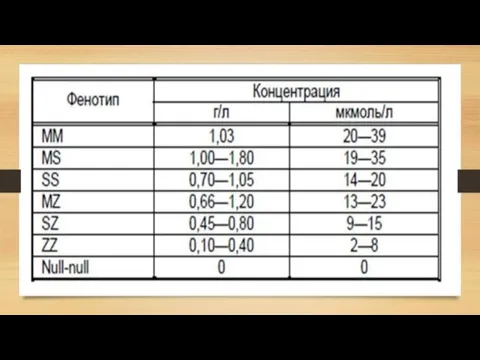
- 14. Риск развития эмфиземы значительно возрастает при снижении уровня сывороточного А1-АТ менее 0,8 г/л (11 ммоль/л) (норма
- 15. Уровень альфа-1-антитриписина в норме и физиологические колебания. Наибольшие количества А1АТ содержатся в сыворотке крови, обеспечивая 90%
- 16. Эмфизема (с греческого – «надувать, разбухать») – заболевание легких, характеризующееся патологическим расширением полостей. Легкое становится объемным
- 17. Клиника Среди основных симптомов дефицита альфа-1-антитрипсина необходимо выделить следующие: одышка, свистящее дыхание и сухие свистящие хрипы
- 18. Диагностика Заболевание диагностируется очень редко. В большинстве случаев, больные альфа-1-антитрипсином безуспешно лечатся с неверными диагнозами —
- 19. Несмотря на довольно широкую распространенность недостаточности А1АТ и несомненные преимущества ее ранней диагностики, лишь в десятке
- 20. Лечение дефицита альфа-1-антитрипсина Специфических методов лечения не существует. Для предупреждения прогрессирования заболевания необходимо избегать неблагоприятного воздействия
- 22. Скачать презентацию
Слайд 3 Наследственная недостаточность α1антитрипсина (α1АТ) – генетическое заболевание, клинически проявляющееся эмфиземой легких,
Наследственная недостаточность α1антитрипсина (α1АТ) – генетическое заболевание, клинически проявляющееся эмфиземой легких,

Слайд 4 Общие сведения об альфа-1-антитриписине
А1АТ представляет собой низкомолекулярный гликопротеин, состоящий из
Общие сведения об альфа-1-антитриписине
А1АТ представляет собой низкомолекулярный гликопротеин, состоящий из

Слайд 5 Генетический полиморфизм альфа-1-антитриписина
За продукцию А1АТ отвечает ген, расположенный на хромосоме
Генетический полиморфизм альфа-1-антитриписина
За продукцию А1АТ отвечает ген, расположенный на хромосоме

Слайд 6 Есть несколько форм и степеней дефицита, которые главным образом зависят от
Есть несколько форм и степеней дефицита, которые главным образом зависят от

Слайд 8Варианты наследования гена альфа-1-антитрипсина
ММ — оба варианта гена здоровы
MS или MZ (PiMS,
Варианты наследования гена альфа-1-антитрипсина
ММ — оба варианта гена здоровы
MS или MZ (PiMS,

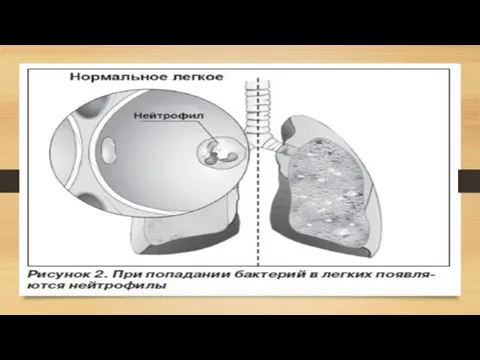
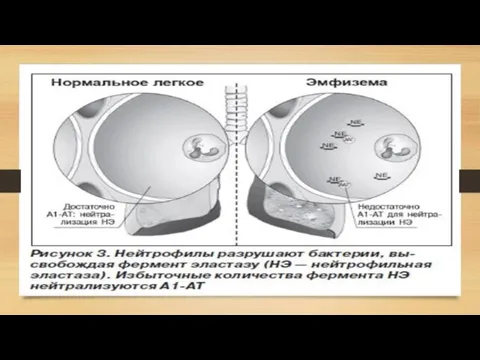

Слайд 14Риск развития эмфиземы значительно возрастает при снижении уровня сывороточного А1-АТ менее 0,8
Риск развития эмфиземы значительно возрастает при снижении уровня сывороточного А1-АТ менее 0,8

Слайд 15 Уровень альфа-1-антитриписина в норме и физиологические колебания.
Наибольшие количества А1АТ содержатся
Уровень альфа-1-антитриписина в норме и физиологические колебания.
Наибольшие количества А1АТ содержатся

Слайд 16 Эмфизема (с греческого – «надувать, разбухать») – заболевание легких, характеризующееся патологическим
Эмфизема (с греческого – «надувать, разбухать») – заболевание легких, характеризующееся патологическим

Слайд 17 Клиника
Среди основных симптомов дефицита альфа-1-антитрипсина необходимо выделить следующие: одышка, свистящее
Клиника
Среди основных симптомов дефицита альфа-1-антитрипсина необходимо выделить следующие: одышка, свистящее

Слайд 18Диагностика
Заболевание диагностируется очень редко. В большинстве случаев, больные альфа-1-антитрипсином безуспешно лечатся
Диагностика
Заболевание диагностируется очень редко. В большинстве случаев, больные альфа-1-антитрипсином безуспешно лечатся

Слайд 19Несмотря на довольно широкую распространенность недостаточности А1АТ и несомненные преимущества ее ранней
Несмотря на довольно широкую распространенность недостаточности А1АТ и несомненные преимущества ее ранней

Слайд 20 Лечение дефицита альфа-1-антитрипсина
Специфических методов лечения не существует. Для предупреждения прогрессирования заболевания
Лечение дефицита альфа-1-антитрипсина
Специфических методов лечения не существует. Для предупреждения прогрессирования заболевания

 Болезнь Паркинсона
Болезнь Паркинсона Мертворождение. Показатель перинатальной смертности. Материнская смерть
Мертворождение. Показатель перинатальной смертности. Материнская смерть Гемодинамика
Гемодинамика Клиническая фармакология лекарственных средств, применяемых при заболеваниях желудочно-кишечного тракта (часть 1)
Клиническая фармакология лекарственных средств, применяемых при заболеваниях желудочно-кишечного тракта (часть 1) Оперативные вмешательства на лимфатической системе
Оперативные вмешательства на лимфатической системе Эпилепсия клиника, диагностика, лечение
Эпилепсия клиника, диагностика, лечение Роль гормонов в обмене веществ, росте и развитии организма
Роль гормонов в обмене веществ, росте и развитии организма О вреде аборта
О вреде аборта Основные виды шока. Травматический и геморрагический шок. Неотложная помощь при шоке
Основные виды шока. Травматический и геморрагический шок. Неотложная помощь при шоке Poisons and allergens of plants and animal origin.their effect on the human body
Poisons and allergens of plants and animal origin.their effect on the human body Анализ эффективности современных методов диагностики и лечения миомы матки
Анализ эффективности современных методов диагностики и лечения миомы матки Первая помощь при воздействии высоких и низких температур
Первая помощь при воздействии высоких и низких температур Пульпа
Пульпа Топ-5 цветов по мнению дальтоников
Топ-5 цветов по мнению дальтоников Физиология крови. Состав крови. Плазма крови. Функции эритроцитов
Физиология крови. Состав крови. Плазма крови. Функции эритроцитов Паллиативная помощь в Бразилии
Паллиативная помощь в Бразилии Анестезиология и интенсивная терапия в эндокринной хирургии
Анестезиология и интенсивная терапия в эндокринной хирургии КДС сұрақ-жауап
КДС сұрақ-жауап Деменция с тельцами Леви
Деменция с тельцами Леви Вторичные головные боли
Вторичные головные боли Жүкті әйелдердегі эндокриндік жүйе қызметі
Жүкті әйелдердегі эндокриндік жүйе қызметі Методы обследования больных с травмами и заболеваниями опорно-двигательного аппарата
Методы обследования больных с травмами и заболеваниями опорно-двигательного аппарата Прямые_и_плоскости_в_архитектуре_и_строительстве_2
Прямые_и_плоскости_в_архитектуре_и_строительстве_2 Исследование шишек сосны обыкновенной как источника дубильных веществ
Исследование шишек сосны обыкновенной как источника дубильных веществ Профилактика инфекций области хирургического вмешательства
Профилактика инфекций области хирургического вмешательства Производные пиримидина
Производные пиримидина Аускультация сердца при клапанных пороках сердца. Трикуспидальные и пульмональные пороки сердца. №13 лекция
Аускультация сердца при клапанных пороках сердца. Трикуспидальные и пульмональные пороки сердца. №13 лекция ХСН. Летняя школа патологии
ХСН. Летняя школа патологии